Faeze Keshavarz-Rahaghi, Erin Pleasance, Steven J M Jones
{"title":"机器学习揭示原发性和转移性肿瘤中癌症相关基因的转录模式。","authors":"Faeze Keshavarz-Rahaghi, Erin Pleasance, Steven J M Jones","doi":"10.1186/s12915-025-02339-z","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>A key to understanding cancer is to determine the impact on the cellular pathways caused by the repertoire of DNA changes accrued in a cancer cell. Exploring the interactions between genomic aberrations and the expressed transcriptome can not only improve our understanding of the disease but also identify potential therapeutic approaches.</p><p><strong>Results: </strong>Using random forest models, we successfully identified transcriptional patterns associated with the loss of wild-type activity in cancer-related genes across various tumour types. While genes like TP53 and CDKN2A exhibited unique pan-cancer transcriptional patterns, others like ATRX, BRAF, and NRAS showed tumour-type-specific expression patterns. We also observed that genes like AR and ERBB4 did not lead to strong detectable patterns in the transcriptome when disrupted. Our investigation has also led to the identification of genes highly associated with transcriptional patterns. For instance, DRG2 emerged as the top contributor in classification of ATRX alterations in lower-grade gliomas and was significantly downregulated in ATRX mutant tumours. Additionally, transcriptional features important in classification of PTEN aberrations, such as CDCA8, AURKA, and CDC20, were found to be closely related to PTEN function.</p><p><strong>Conclusions: </strong>Our findings demonstrate the utility of machine learning in interpretation of cancer genomic data and provide new avenues for development of targeted therapies tailored to individual patients with cancer. Our analysis on the transcriptome revealed genes with expression levels strongly correlated with alterations in cancer-related genes. Additionally, we identified AURKA inhibitors as potential therapeutic option for tumours with alterations in tumour suppressors like FBXW7 or NSD1.</p>","PeriodicalId":9339,"journal":{"name":"BMC Biology","volume":"23 1","pages":"246"},"PeriodicalIF":4.5000,"publicationDate":"2025-08-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12329921/pdf/","citationCount":"0","resultStr":"{\"title\":\"Transcriptional patterns of cancer-related genes in primary and metastatic tumours revealed by machine learning.\",\"authors\":\"Faeze Keshavarz-Rahaghi, Erin Pleasance, Steven J M Jones\",\"doi\":\"10.1186/s12915-025-02339-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>A key to understanding cancer is to determine the impact on the cellular pathways caused by the repertoire of DNA changes accrued in a cancer cell. Exploring the interactions between genomic aberrations and the expressed transcriptome can not only improve our understanding of the disease but also identify potential therapeutic approaches.</p><p><strong>Results: </strong>Using random forest models, we successfully identified transcriptional patterns associated with the loss of wild-type activity in cancer-related genes across various tumour types. While genes like TP53 and CDKN2A exhibited unique pan-cancer transcriptional patterns, others like ATRX, BRAF, and NRAS showed tumour-type-specific expression patterns. We also observed that genes like AR and ERBB4 did not lead to strong detectable patterns in the transcriptome when disrupted. Our investigation has also led to the identification of genes highly associated with transcriptional patterns. For instance, DRG2 emerged as the top contributor in classification of ATRX alterations in lower-grade gliomas and was significantly downregulated in ATRX mutant tumours. Additionally, transcriptional features important in classification of PTEN aberrations, such as CDCA8, AURKA, and CDC20, were found to be closely related to PTEN function.</p><p><strong>Conclusions: </strong>Our findings demonstrate the utility of machine learning in interpretation of cancer genomic data and provide new avenues for development of targeted therapies tailored to individual patients with cancer. Our analysis on the transcriptome revealed genes with expression levels strongly correlated with alterations in cancer-related genes. Additionally, we identified AURKA inhibitors as potential therapeutic option for tumours with alterations in tumour suppressors like FBXW7 or NSD1.</p>\",\"PeriodicalId\":9339,\"journal\":{\"name\":\"BMC Biology\",\"volume\":\"23 1\",\"pages\":\"246\"},\"PeriodicalIF\":4.5000,\"publicationDate\":\"2025-08-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12329921/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s12915-025-02339-z\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12915-025-02339-z","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOLOGY","Score":null,"Total":0}
Transcriptional patterns of cancer-related genes in primary and metastatic tumours revealed by machine learning.
Background: A key to understanding cancer is to determine the impact on the cellular pathways caused by the repertoire of DNA changes accrued in a cancer cell. Exploring the interactions between genomic aberrations and the expressed transcriptome can not only improve our understanding of the disease but also identify potential therapeutic approaches.
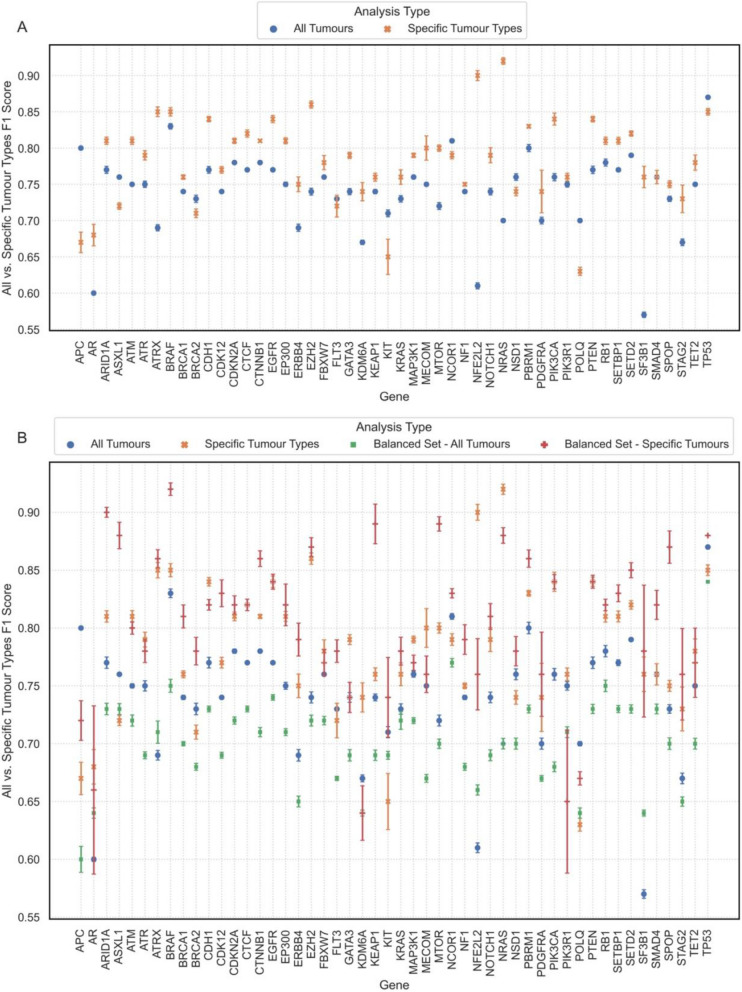
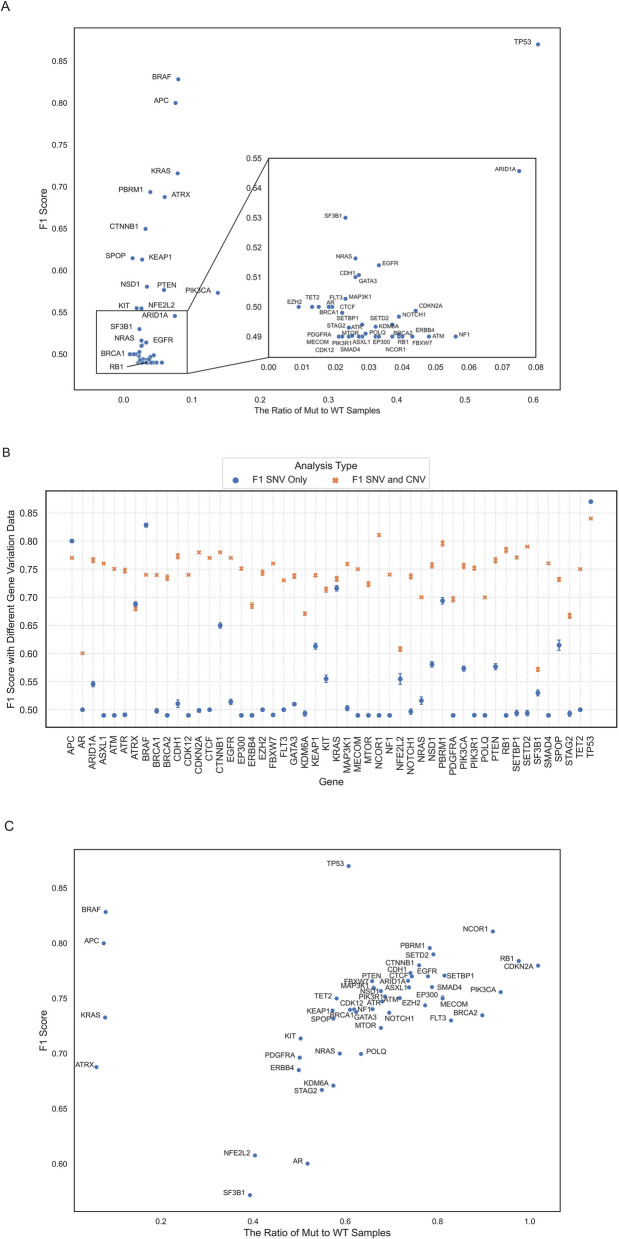
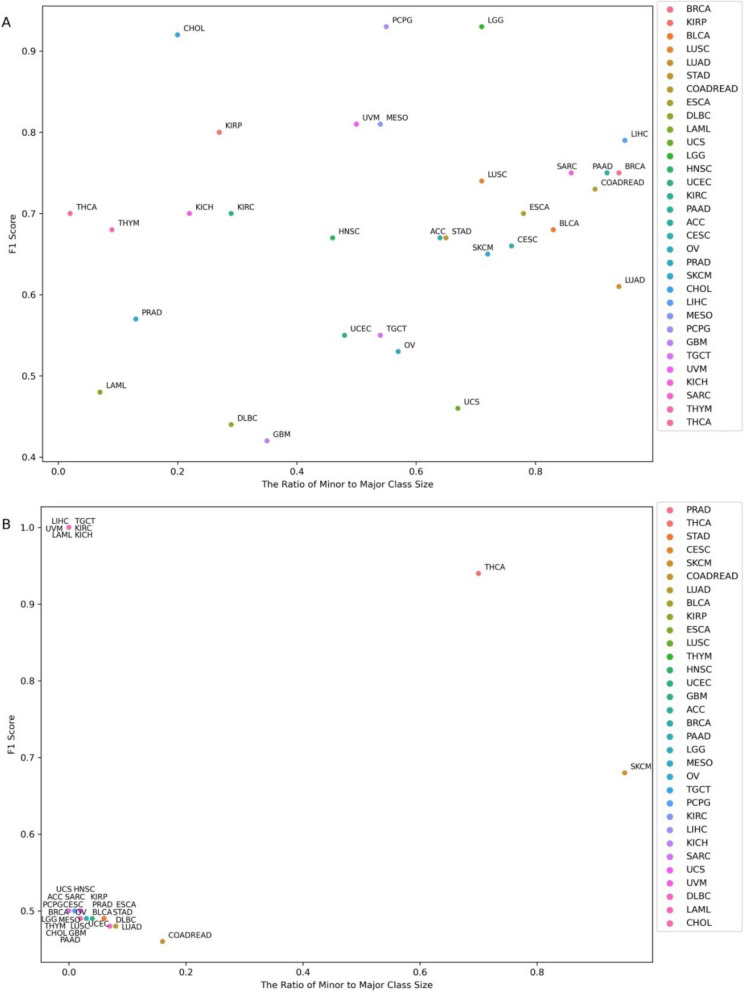
Results: Using random forest models, we successfully identified transcriptional patterns associated with the loss of wild-type activity in cancer-related genes across various tumour types. While genes like TP53 and CDKN2A exhibited unique pan-cancer transcriptional patterns, others like ATRX, BRAF, and NRAS showed tumour-type-specific expression patterns. We also observed that genes like AR and ERBB4 did not lead to strong detectable patterns in the transcriptome when disrupted. Our investigation has also led to the identification of genes highly associated with transcriptional patterns. For instance, DRG2 emerged as the top contributor in classification of ATRX alterations in lower-grade gliomas and was significantly downregulated in ATRX mutant tumours. Additionally, transcriptional features important in classification of PTEN aberrations, such as CDCA8, AURKA, and CDC20, were found to be closely related to PTEN function.
Conclusions: Our findings demonstrate the utility of machine learning in interpretation of cancer genomic data and provide new avenues for development of targeted therapies tailored to individual patients with cancer. Our analysis on the transcriptome revealed genes with expression levels strongly correlated with alterations in cancer-related genes. Additionally, we identified AURKA inhibitors as potential therapeutic option for tumours with alterations in tumour suppressors like FBXW7 or NSD1.
期刊介绍:
BMC Biology is a broad scope journal covering all areas of biology. Our content includes research articles, new methods and tools. BMC Biology also publishes reviews, Q&A, and commentaries.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


