{"title":"携带ABCG5和HBA基因突变的谷固醇血症:一例报告和文献回顾。","authors":"Xiaobing Sun, Jiong Wu, Pu Chen, Ruiqing He, Ting Li, Qingwen Zeng, Qi Hou","doi":"10.1186/s13256-025-05439-0","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Mutations in the ABCG5 gene can cause sitosterolemia, which is a rare lipid metabolism disorder characterized by impaired regulation of phytosterols, leading to their excessive accumulation in tissues and organs, which triggers various complications. If left untreated, it may cause serious issues, often presenting first as xanthomas on the skin and other tissues.</p><p><strong>Case presentation: </strong>A 9-year-old female Chinese Zhuang patient developed her first xanthomas on her knees at the age of 4, which progressively spread across her body over the years. Initial blood tests revealed elevated plasma cholesterol and low-density lipoprotein, and she was misdiagnosed with familial hypercholesterolemia, leading to ineffective treatment. Despite visiting several hospitals, the underlying cause remained unidentified, and the patient was eventually admitted to our hospital for further evaluation. The complete blood count showed mild hypochromic microcytic anemia and blood smears showed microcytic hypochromic anemia and the presence of giant platelets in the peripheral blood. Plasma phytosterol profiling revealed significantly elevated phytosterol levels, and whole exome sequencing detected a homozygous mutation in the ABCG5 gene (c.751C > T, p.Q251*). On the basis of these findings, the patient was diagnosed with sitosterolemia. Her parents and younger brother were found to carry the heterozygous mutation but exhibited no clinical symptoms. In addition, iron metabolism tests and DNA copy number multidetection technology, along with single nucleotide polymorphism typing, revealed that the patient also had a silent alpha-thalassemia trait (genotype: HBA, -α3.7/αα).</p><p><strong>Conclusion: </strong>Sitosterolemia is a rare lipid metabolism disorder that should be considered in patients presenting with multiple xanthomas, severe hypercholesterolemia, or elevated low-density lipoprotein-cholesterol levels. Diagnosis can be confirmed through phytosterol detection and molecular testing. Early diagnosis allows for dietary recommendations-such as restricting cholesterol and phytosterol intake-and, if necessary, treatment with medications such as ezetimibe. As we know, alpha-thalassemia is able to cause microcytosis and phytosterolemia may cause stomatocytosis in peripheral blood. However, there are no reports of two gene mutations occurring simultaneously in the same individual, and no stomatocytosis was observed in our patient. Hence, this suggests that the mutual regulation of two diseases and the effects on red blood cell membranes may exist, and the underlying mechanisms of this phenomenon are valuable for further research.</p>","PeriodicalId":16236,"journal":{"name":"Journal of Medical Case Reports","volume":"19 1","pages":"387"},"PeriodicalIF":0.8000,"publicationDate":"2025-08-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12323034/pdf/","citationCount":"0","resultStr":"{\"title\":\"Sitosterolemia carrying both ABCG5 and HBA gene mutations: a case report and review of the literature.\",\"authors\":\"Xiaobing Sun, Jiong Wu, Pu Chen, Ruiqing He, Ting Li, Qingwen Zeng, Qi Hou\",\"doi\":\"10.1186/s13256-025-05439-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Mutations in the ABCG5 gene can cause sitosterolemia, which is a rare lipid metabolism disorder characterized by impaired regulation of phytosterols, leading to their excessive accumulation in tissues and organs, which triggers various complications. If left untreated, it may cause serious issues, often presenting first as xanthomas on the skin and other tissues.</p><p><strong>Case presentation: </strong>A 9-year-old female Chinese Zhuang patient developed her first xanthomas on her knees at the age of 4, which progressively spread across her body over the years. Initial blood tests revealed elevated plasma cholesterol and low-density lipoprotein, and she was misdiagnosed with familial hypercholesterolemia, leading to ineffective treatment. Despite visiting several hospitals, the underlying cause remained unidentified, and the patient was eventually admitted to our hospital for further evaluation. The complete blood count showed mild hypochromic microcytic anemia and blood smears showed microcytic hypochromic anemia and the presence of giant platelets in the peripheral blood. Plasma phytosterol profiling revealed significantly elevated phytosterol levels, and whole exome sequencing detected a homozygous mutation in the ABCG5 gene (c.751C > T, p.Q251*). On the basis of these findings, the patient was diagnosed with sitosterolemia. Her parents and younger brother were found to carry the heterozygous mutation but exhibited no clinical symptoms. In addition, iron metabolism tests and DNA copy number multidetection technology, along with single nucleotide polymorphism typing, revealed that the patient also had a silent alpha-thalassemia trait (genotype: HBA, -α3.7/αα).</p><p><strong>Conclusion: </strong>Sitosterolemia is a rare lipid metabolism disorder that should be considered in patients presenting with multiple xanthomas, severe hypercholesterolemia, or elevated low-density lipoprotein-cholesterol levels. Diagnosis can be confirmed through phytosterol detection and molecular testing. Early diagnosis allows for dietary recommendations-such as restricting cholesterol and phytosterol intake-and, if necessary, treatment with medications such as ezetimibe. As we know, alpha-thalassemia is able to cause microcytosis and phytosterolemia may cause stomatocytosis in peripheral blood. However, there are no reports of two gene mutations occurring simultaneously in the same individual, and no stomatocytosis was observed in our patient. Hence, this suggests that the mutual regulation of two diseases and the effects on red blood cell membranes may exist, and the underlying mechanisms of this phenomenon are valuable for further research.</p>\",\"PeriodicalId\":16236,\"journal\":{\"name\":\"Journal of Medical Case Reports\",\"volume\":\"19 1\",\"pages\":\"387\"},\"PeriodicalIF\":0.8000,\"publicationDate\":\"2025-08-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12323034/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Medical Case Reports\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s13256-025-05439-0\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"MEDICINE, GENERAL & INTERNAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Medical Case Reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s13256-025-05439-0","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
Sitosterolemia carrying both ABCG5 and HBA gene mutations: a case report and review of the literature.
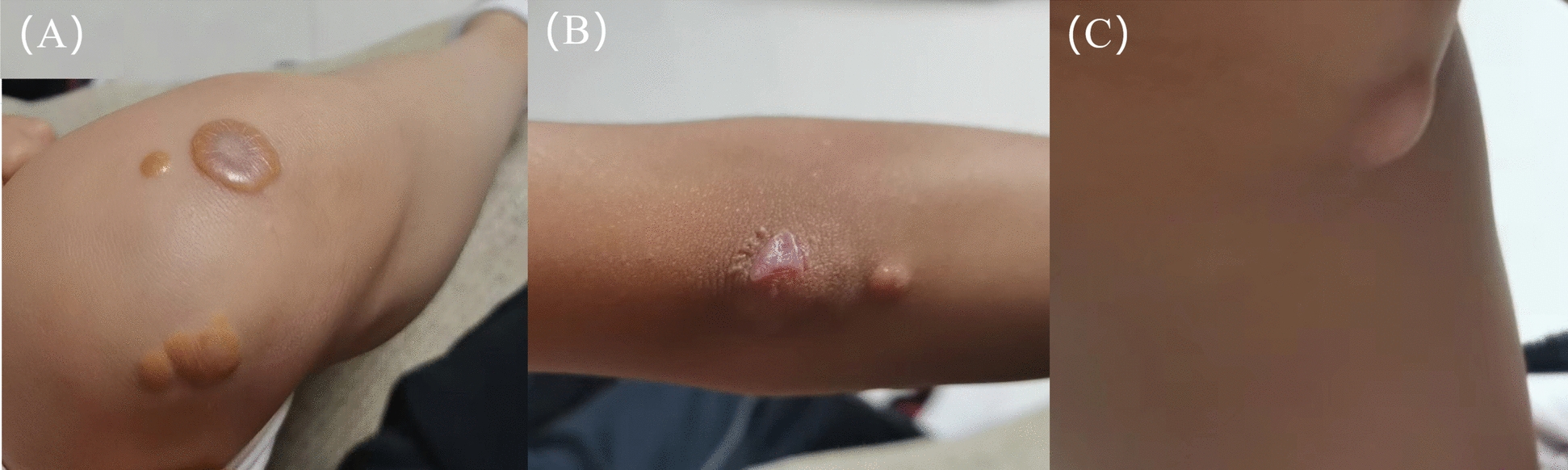
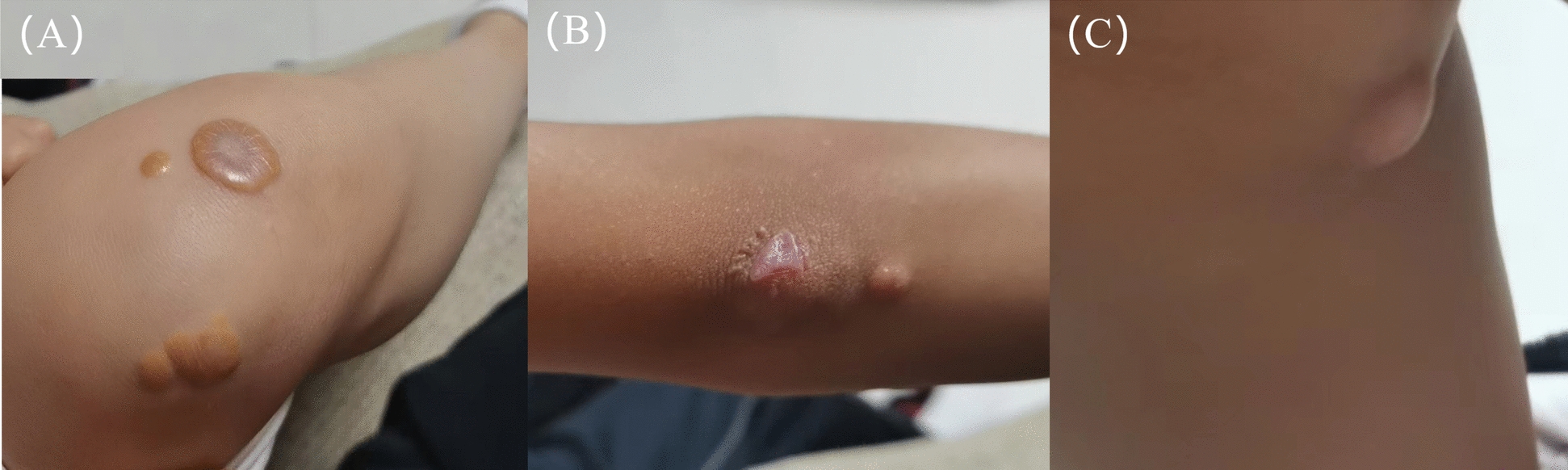
Background: Mutations in the ABCG5 gene can cause sitosterolemia, which is a rare lipid metabolism disorder characterized by impaired regulation of phytosterols, leading to their excessive accumulation in tissues and organs, which triggers various complications. If left untreated, it may cause serious issues, often presenting first as xanthomas on the skin and other tissues.
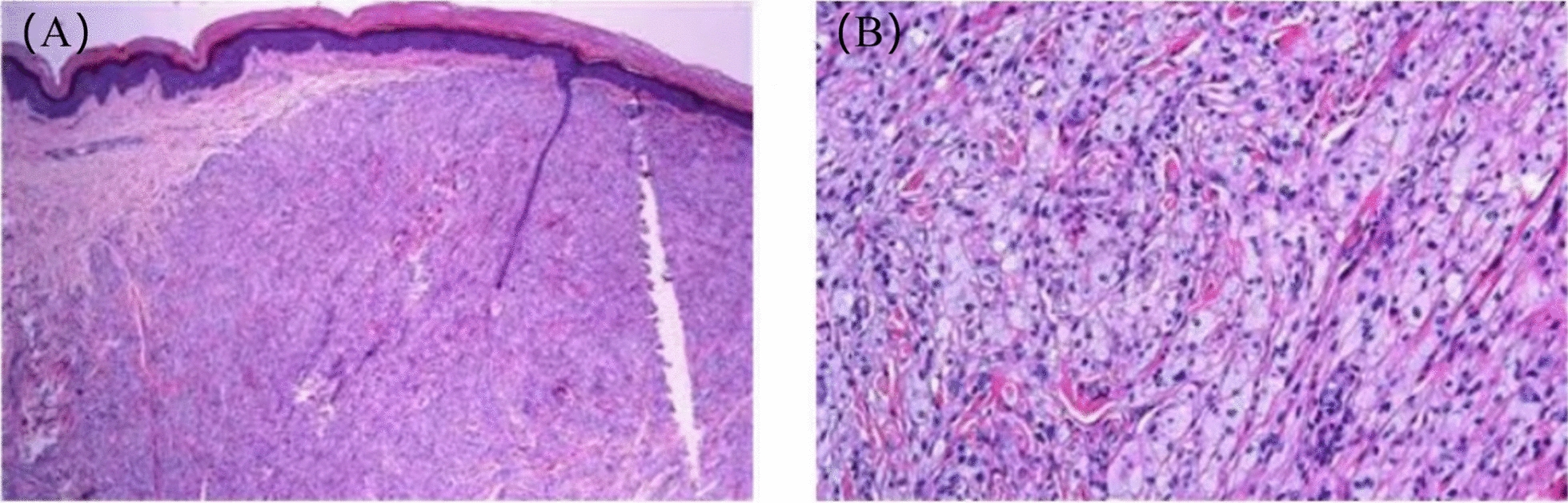
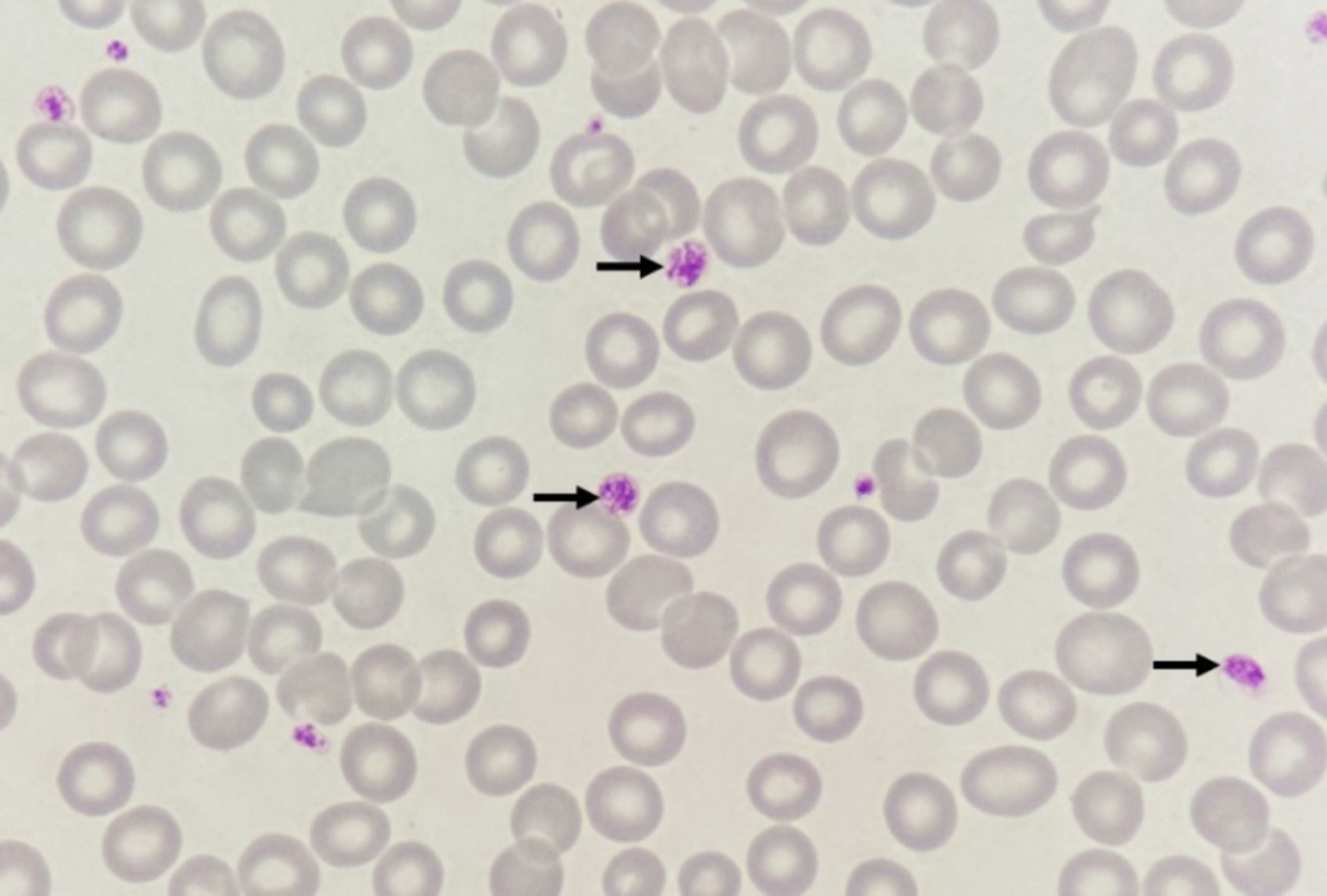
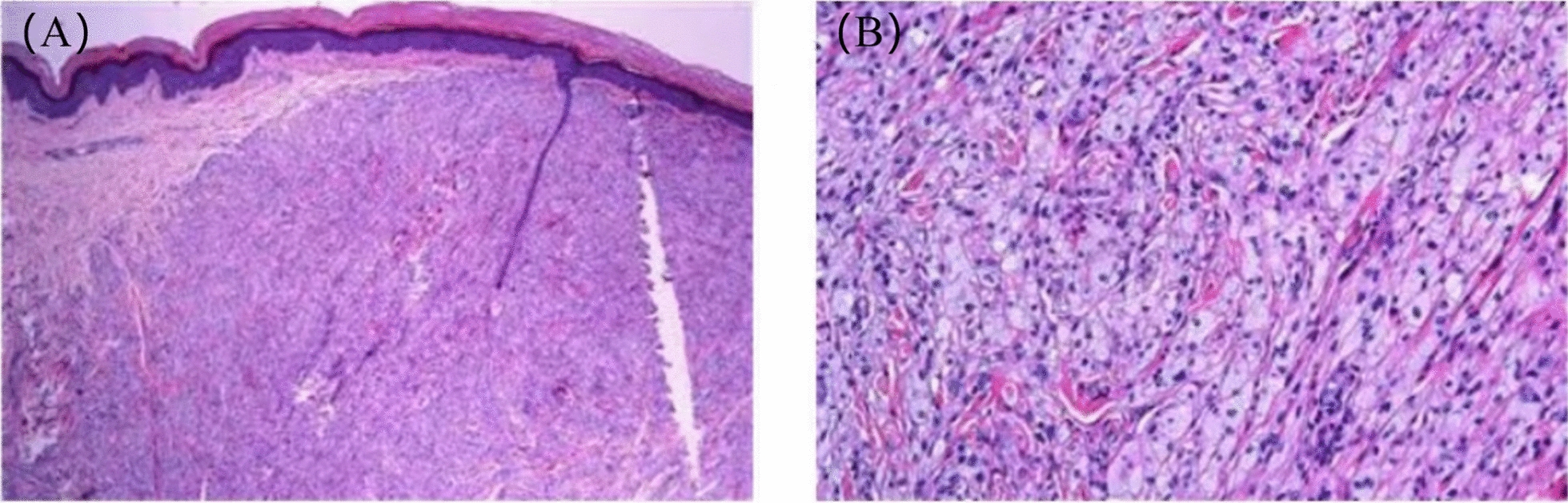
Case presentation: A 9-year-old female Chinese Zhuang patient developed her first xanthomas on her knees at the age of 4, which progressively spread across her body over the years. Initial blood tests revealed elevated plasma cholesterol and low-density lipoprotein, and she was misdiagnosed with familial hypercholesterolemia, leading to ineffective treatment. Despite visiting several hospitals, the underlying cause remained unidentified, and the patient was eventually admitted to our hospital for further evaluation. The complete blood count showed mild hypochromic microcytic anemia and blood smears showed microcytic hypochromic anemia and the presence of giant platelets in the peripheral blood. Plasma phytosterol profiling revealed significantly elevated phytosterol levels, and whole exome sequencing detected a homozygous mutation in the ABCG5 gene (c.751C > T, p.Q251*). On the basis of these findings, the patient was diagnosed with sitosterolemia. Her parents and younger brother were found to carry the heterozygous mutation but exhibited no clinical symptoms. In addition, iron metabolism tests and DNA copy number multidetection technology, along with single nucleotide polymorphism typing, revealed that the patient also had a silent alpha-thalassemia trait (genotype: HBA, -α3.7/αα).
Conclusion: Sitosterolemia is a rare lipid metabolism disorder that should be considered in patients presenting with multiple xanthomas, severe hypercholesterolemia, or elevated low-density lipoprotein-cholesterol levels. Diagnosis can be confirmed through phytosterol detection and molecular testing. Early diagnosis allows for dietary recommendations-such as restricting cholesterol and phytosterol intake-and, if necessary, treatment with medications such as ezetimibe. As we know, alpha-thalassemia is able to cause microcytosis and phytosterolemia may cause stomatocytosis in peripheral blood. However, there are no reports of two gene mutations occurring simultaneously in the same individual, and no stomatocytosis was observed in our patient. Hence, this suggests that the mutual regulation of two diseases and the effects on red blood cell membranes may exist, and the underlying mechanisms of this phenomenon are valuable for further research.
期刊介绍:
JMCR is an open access, peer-reviewed online journal that will consider any original case report that expands the field of general medical knowledge. Reports should show one of the following: 1. Unreported or unusual side effects or adverse interactions involving medications 2. Unexpected or unusual presentations of a disease 3. New associations or variations in disease processes 4. Presentations, diagnoses and/or management of new and emerging diseases 5. An unexpected association between diseases or symptoms 6. An unexpected event in the course of observing or treating a patient 7. Findings that shed new light on the possible pathogenesis of a disease or an adverse effect

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


