{"title":"通过保持嵌入之间的原始拓扑关系来准确预测药物-蛋白质相互作用。","authors":"Yanfei Li, Xiran Chen, Shuqin Wang, Jinmao Wei","doi":"10.1186/s12915-025-02338-0","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Learning-based methods have recently demonstrated strong potential in predicting drug-protein interactions (DPIs). However, existing approaches often fail to achieve accurate predictions on real-world imbalanced datasets while maintaining high generalizability and scalability, limiting their practical applicability.</p><p><strong>Results: </strong>This study proposes a highly generalized model, GLDPI, aimed at improving prediction accuracy in imbalanced scenarios by preserving the topological relationships among initial molecular representations in the embedding space. Specifically, GLDPI employs dedicated encoders to transform one-dimensional sequence information of drugs and proteins into embedding representations and efficiently calculates the likelihood of DPIs using cosine similarity. Additionally, we introduce a prior loss function based on the guilt-by-association principle to ensure that the topology of the embedding space aligns with the structure of the initial drug-protein network. This design enables GLDPI to effectively capture network relationships and key features of molecular interactions, thereby significantly enhancing predictive performance.</p><p><strong>Conclusions: </strong>Experimental results highlight GLDPI's superior performance on multiple highly imbalanced benchmark datasets, achieving over a 100% improvement in the AUPR metric compared to state-of-the-art methods. Additionally, GLDPI demonstrates exceptional generalization capabilities in cold-start experiments, excelling in predicting novel drug-protein interactions. Furthermore, the model exhibits remarkable scalability, efficiently inferring approximately <math><mrow><mn>1.2</mn> <mo>×</mo> <msup><mn>10</mn> <mn>10</mn></msup> </mrow> </math> drug-protein pairs in less than 10 h.</p>","PeriodicalId":9339,"journal":{"name":"BMC Biology","volume":"23 1","pages":"243"},"PeriodicalIF":4.5000,"publicationDate":"2025-08-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12326672/pdf/","citationCount":"0","resultStr":"{\"title\":\"Accurate prediction of drug-protein interactions by maintaining the original topological relationships among embeddings.\",\"authors\":\"Yanfei Li, Xiran Chen, Shuqin Wang, Jinmao Wei\",\"doi\":\"10.1186/s12915-025-02338-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Learning-based methods have recently demonstrated strong potential in predicting drug-protein interactions (DPIs). However, existing approaches often fail to achieve accurate predictions on real-world imbalanced datasets while maintaining high generalizability and scalability, limiting their practical applicability.</p><p><strong>Results: </strong>This study proposes a highly generalized model, GLDPI, aimed at improving prediction accuracy in imbalanced scenarios by preserving the topological relationships among initial molecular representations in the embedding space. Specifically, GLDPI employs dedicated encoders to transform one-dimensional sequence information of drugs and proteins into embedding representations and efficiently calculates the likelihood of DPIs using cosine similarity. Additionally, we introduce a prior loss function based on the guilt-by-association principle to ensure that the topology of the embedding space aligns with the structure of the initial drug-protein network. This design enables GLDPI to effectively capture network relationships and key features of molecular interactions, thereby significantly enhancing predictive performance.</p><p><strong>Conclusions: </strong>Experimental results highlight GLDPI's superior performance on multiple highly imbalanced benchmark datasets, achieving over a 100% improvement in the AUPR metric compared to state-of-the-art methods. Additionally, GLDPI demonstrates exceptional generalization capabilities in cold-start experiments, excelling in predicting novel drug-protein interactions. Furthermore, the model exhibits remarkable scalability, efficiently inferring approximately <math><mrow><mn>1.2</mn> <mo>×</mo> <msup><mn>10</mn> <mn>10</mn></msup> </mrow> </math> drug-protein pairs in less than 10 h.</p>\",\"PeriodicalId\":9339,\"journal\":{\"name\":\"BMC Biology\",\"volume\":\"23 1\",\"pages\":\"243\"},\"PeriodicalIF\":4.5000,\"publicationDate\":\"2025-08-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12326672/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s12915-025-02338-0\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12915-025-02338-0","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOLOGY","Score":null,"Total":0}
Accurate prediction of drug-protein interactions by maintaining the original topological relationships among embeddings.
Background: Learning-based methods have recently demonstrated strong potential in predicting drug-protein interactions (DPIs). However, existing approaches often fail to achieve accurate predictions on real-world imbalanced datasets while maintaining high generalizability and scalability, limiting their practical applicability.
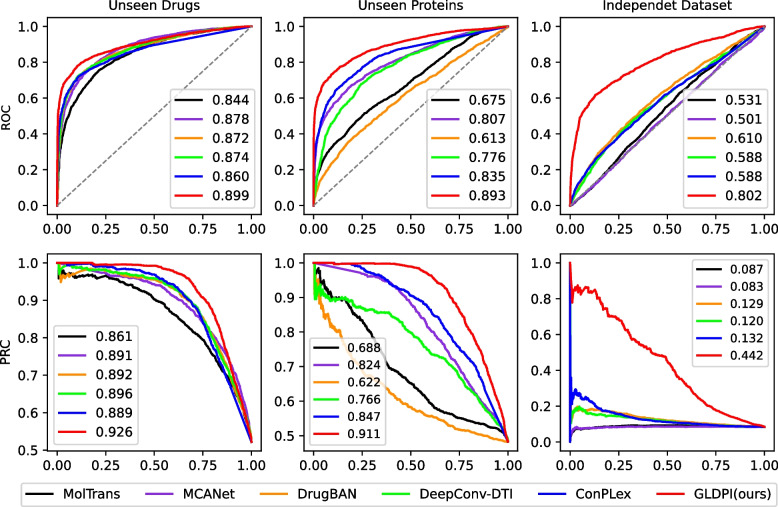
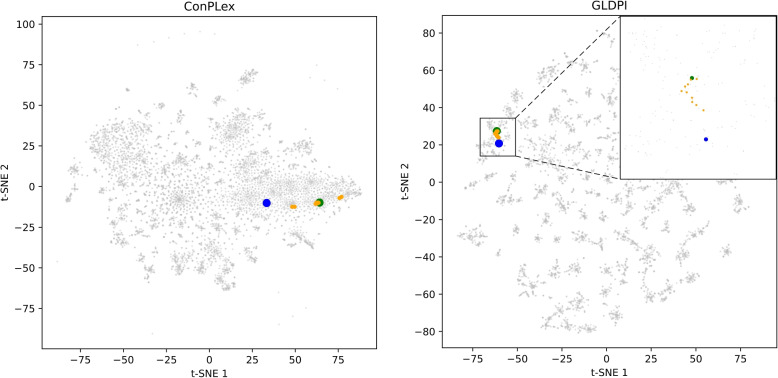
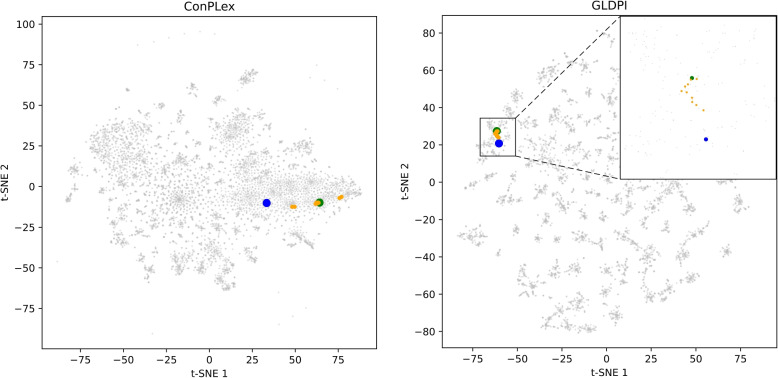
Results: This study proposes a highly generalized model, GLDPI, aimed at improving prediction accuracy in imbalanced scenarios by preserving the topological relationships among initial molecular representations in the embedding space. Specifically, GLDPI employs dedicated encoders to transform one-dimensional sequence information of drugs and proteins into embedding representations and efficiently calculates the likelihood of DPIs using cosine similarity. Additionally, we introduce a prior loss function based on the guilt-by-association principle to ensure that the topology of the embedding space aligns with the structure of the initial drug-protein network. This design enables GLDPI to effectively capture network relationships and key features of molecular interactions, thereby significantly enhancing predictive performance.
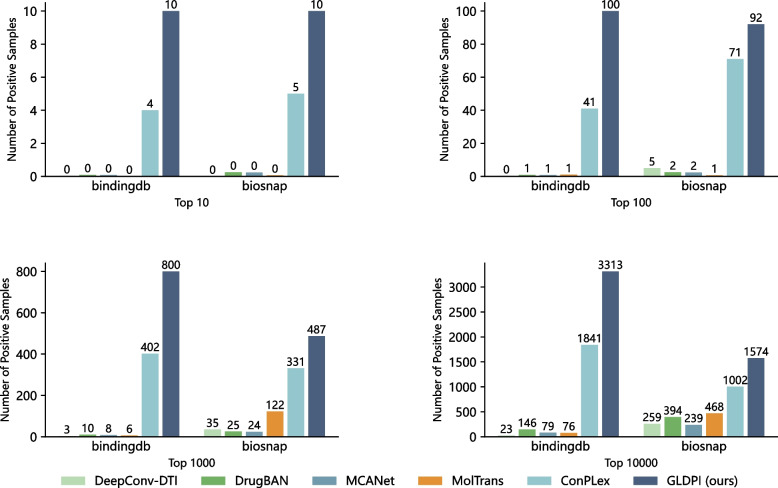
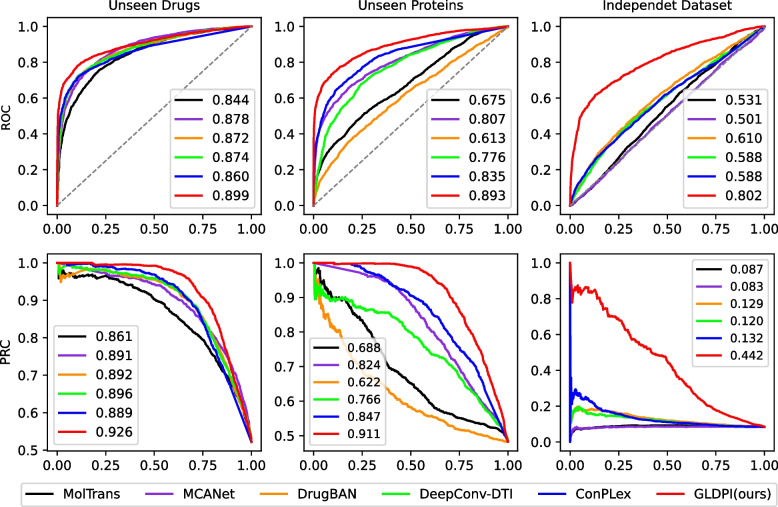
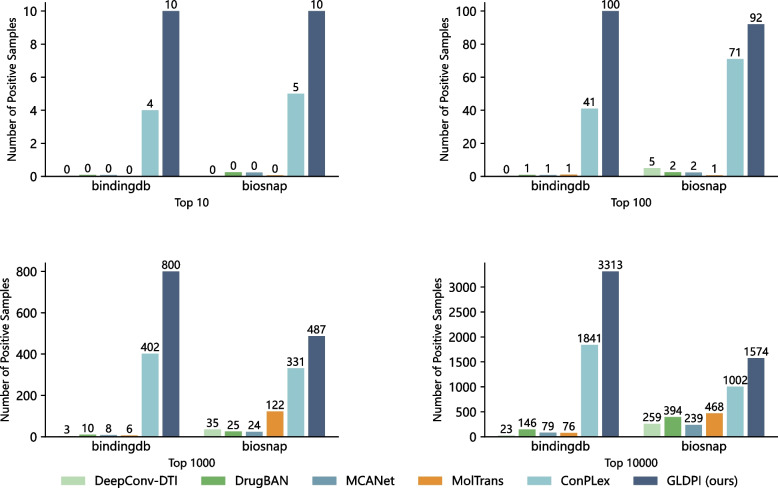
Conclusions: Experimental results highlight GLDPI's superior performance on multiple highly imbalanced benchmark datasets, achieving over a 100% improvement in the AUPR metric compared to state-of-the-art methods. Additionally, GLDPI demonstrates exceptional generalization capabilities in cold-start experiments, excelling in predicting novel drug-protein interactions. Furthermore, the model exhibits remarkable scalability, efficiently inferring approximately drug-protein pairs in less than 10 h.
期刊介绍:
BMC Biology is a broad scope journal covering all areas of biology. Our content includes research articles, new methods and tools. BMC Biology also publishes reviews, Q&A, and commentaries.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


