{"title":"染色体1p31.1缺失:发育迟缓、张力低下、隐睾、口系带异常、足部畸形1例。","authors":"Tatiana Mikhailova, Ria Garg","doi":"10.1155/crig/6152118","DOIUrl":null,"url":null,"abstract":"<p><p>Deletions within the chromosomal locus 1p31.1 are rare, with only a limited number of documented cases. The typical clinical presentation includes intellectual disability, failure to thrive, and craniofacial abnormalities. Some cases may also present with cardiac, gastrointestinal, and genitourinary malformations. Variability in deletion size contributes to a broad spectrum of clinical phenotypes, and a comprehensive understanding of the syndrome's manifestations is still evolving. This case study aims to provide additional insights into 1p31.1 microdeletion syndrome, enhancing knowledge of its genetic and phenotypic characteristics to improve recognition by clinicians. Here, we report a case featuring a 14.385 Mb deletion isolated to the 1p31.1 region, encompassing 41 genes. The deletion manifested with microcephaly, distinctive facial morphology, hypotonia, developmental delay, bilateral cryptorchidism, and flat feet. Notably, our case also exhibited congenital thickening of the lingual and labial frenulum, a trait not typically associated with this deletion.</p>","PeriodicalId":30325,"journal":{"name":"Case Reports in Genetics","volume":"2025 ","pages":"6152118"},"PeriodicalIF":0.0000,"publicationDate":"2025-07-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12318624/pdf/","citationCount":"0","resultStr":"{\"title\":\"Chromosome 1p31.1 Deletion: A Case With Developmental Delay, Hypotonia, Cryptorchidism, Abnormal Oral Frenulum, and Feet Deformity.\",\"authors\":\"Tatiana Mikhailova, Ria Garg\",\"doi\":\"10.1155/crig/6152118\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Deletions within the chromosomal locus 1p31.1 are rare, with only a limited number of documented cases. The typical clinical presentation includes intellectual disability, failure to thrive, and craniofacial abnormalities. Some cases may also present with cardiac, gastrointestinal, and genitourinary malformations. Variability in deletion size contributes to a broad spectrum of clinical phenotypes, and a comprehensive understanding of the syndrome's manifestations is still evolving. This case study aims to provide additional insights into 1p31.1 microdeletion syndrome, enhancing knowledge of its genetic and phenotypic characteristics to improve recognition by clinicians. Here, we report a case featuring a 14.385 Mb deletion isolated to the 1p31.1 region, encompassing 41 genes. The deletion manifested with microcephaly, distinctive facial morphology, hypotonia, developmental delay, bilateral cryptorchidism, and flat feet. Notably, our case also exhibited congenital thickening of the lingual and labial frenulum, a trait not typically associated with this deletion.</p>\",\"PeriodicalId\":30325,\"journal\":{\"name\":\"Case Reports in Genetics\",\"volume\":\"2025 \",\"pages\":\"6152118\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2025-07-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12318624/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Case Reports in Genetics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/crig/6152118\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Genetics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/crig/6152118","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Chromosome 1p31.1 Deletion: A Case With Developmental Delay, Hypotonia, Cryptorchidism, Abnormal Oral Frenulum, and Feet Deformity.
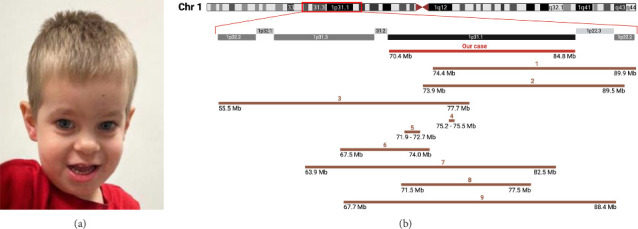
Deletions within the chromosomal locus 1p31.1 are rare, with only a limited number of documented cases. The typical clinical presentation includes intellectual disability, failure to thrive, and craniofacial abnormalities. Some cases may also present with cardiac, gastrointestinal, and genitourinary malformations. Variability in deletion size contributes to a broad spectrum of clinical phenotypes, and a comprehensive understanding of the syndrome's manifestations is still evolving. This case study aims to provide additional insights into 1p31.1 microdeletion syndrome, enhancing knowledge of its genetic and phenotypic characteristics to improve recognition by clinicians. Here, we report a case featuring a 14.385 Mb deletion isolated to the 1p31.1 region, encompassing 41 genes. The deletion manifested with microcephaly, distinctive facial morphology, hypotonia, developmental delay, bilateral cryptorchidism, and flat feet. Notably, our case also exhibited congenital thickening of the lingual and labial frenulum, a trait not typically associated with this deletion.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


