计划数据独立采集质谱法用于全球蛋白质组学和接近标记
IF 1.7
3区 化学
Q3 PHYSICS, ATOMIC, MOLECULAR & CHEMICAL
引用次数: 0
摘要
基于质谱(MS)的蛋白质组学经常面临冗余或信息不丰富的串联质谱的挑战。虽然数据独立采集(DIA)质谱提供了出色的再现性,但DIA中使用的宽隔离窗口不可避免地产生了大量无用的MS/MS光谱,并损害了肽分析的特异性。在这里,我们探索了在之前的数据依赖获取(DDA)调查运行中确定的有用肽上专门安排DIA的可能性。与传统的静态DIA方法相比,建立并优化了一种schedule -DIA方法,以减少占空比,提高蛋白质的鉴定和定量。我们将Scheduled-DIA方法应用于全局标记和接近标记蛋白质组学。溶酶体接近标记实验尤其受益于Scheduled-DIA方法,该方法增加了鉴定和定量关键溶酶体膜蛋白和溶酶体相互作用物的敏感性。综上所述,Scheduled-DIA是获取高质量蛋白质组学DIA数据的另一种有效策略。本文章由计算机程序翻译,如有差异,请以英文原文为准。
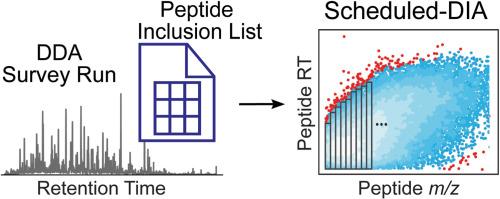
Scheduled data-independent acquisition mass spectrometry for global proteomics and proximity labeling
Mass spectrometry (MS)-based proteomics often faces challenges with redundant or uninformative tandem MS/MS spectra. Although data-independent acquisition (DIA) MS offers excellent reproducibility, the wide isolation windows used in DIA inevitably generate numerous MS/MS spectra that are not useful and compromise the specificity of peptide analysis. Here, we explored the possibility of scheduling DIA exclusively on useful peptides identified in a preceding data-dependent acquisition (DDA) survey run. We established and optimized a Scheduled-DIA method to reduce duty cycle and improve protein identification and quantification compared to the traditional static DIA method. We applied the Scheduled-DIA method to both global and proximity labeling proteomics. Lysosomal proximity labeling experiments particularly benefited from the Scheduled-DIA method, which increased sensitivity for identifying and quantifying key lysosomal membrane proteins and lysosomal interactors. To summarize, Scheduled-DIA is an alternative and useful strategy for acquiring high-quality DIA data for proteomics.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
3.60
自引率
5.60%
发文量
145
审稿时长
71 days
期刊介绍:
The journal invites papers that advance the field of mass spectrometry by exploring fundamental aspects of ion processes using both the experimental and theoretical approaches, developing new instrumentation and experimental strategies for chemical analysis using mass spectrometry, developing new computational strategies for data interpretation and integration, reporting new applications of mass spectrometry and hyphenated techniques in biology, chemistry, geology, and physics.
Papers, in which standard mass spectrometry techniques are used for analysis will not be considered.
IJMS publishes full-length articles, short communications, reviews, and feature articles including young scientist features.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


