Leonie Wenson, Johan Heldin, Marcel Martin, Yücel Erbilgin, Barış Salman, Anders Sundqvist, Wesley Schaal, Friederike A. Sandbaumhüter, Erik T. Jansson, Xingqi Chen, Anton Davidsson, Bo Stenerlöw, Jaime A. Espinoza, Mikael Lindström, Johan Lennartsson, Ola Spjuth, Ola Söderberg
{"title":"通过序列模板错误DNA聚合酶末端标记精确定位单链DNA断裂","authors":"Leonie Wenson, Johan Heldin, Marcel Martin, Yücel Erbilgin, Barış Salman, Anders Sundqvist, Wesley Schaal, Friederike A. Sandbaumhüter, Erik T. Jansson, Xingqi Chen, Anton Davidsson, Bo Stenerlöw, Jaime A. Espinoza, Mikael Lindström, Johan Lennartsson, Ola Spjuth, Ola Söderberg","doi":"10.1038/s41467-025-62512-4","DOIUrl":null,"url":null,"abstract":"<p>The ability to analyze whether DNA contains lesions is essential in identifying mutagenic substances. Currently, the detection of single-stranded DNA breaks (SSBs) lacks precision. To address this limitation, we develop a method for sequence-templated erroneous end-labelling sequencing (STEEL-seq), which enables the mapping of SSBs. The method requires a highly error-prone DNA polymerase, so we engineer a chimeric DNA polymerase, Sloppymerase, capable of replicating DNA in the absence of one nucleotide. Following the omission of a specific nucleotide (e.g., dATP) from the reaction mixture, Sloppymerase introduces mismatches directly downstream of SSBs at positions where deoxyadenosine should occur. This mismatch pattern, coupled with the retention of sequence information flanking these sites, ensures that the identified hits are bona fide SSBs. STEEL-seq is compatible with a variety of sequencing technologies, as demonstrated using Sanger, Illumina, PacBio, and Nanopore systems. Using STEEL-seq, we determine the SSB/base pair frequency in the human genome to range between 0.7 and 3.8 × 10<sup>−6</sup> with an enrichment in active promoter regions.</p>","PeriodicalId":19066,"journal":{"name":"Nature Communications","volume":"19 1","pages":""},"PeriodicalIF":15.7000,"publicationDate":"2025-08-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Precise mapping of single-stranded DNA breaks by sequence-templated erroneous DNA polymerase end-labelling\",\"authors\":\"Leonie Wenson, Johan Heldin, Marcel Martin, Yücel Erbilgin, Barış Salman, Anders Sundqvist, Wesley Schaal, Friederike A. Sandbaumhüter, Erik T. Jansson, Xingqi Chen, Anton Davidsson, Bo Stenerlöw, Jaime A. Espinoza, Mikael Lindström, Johan Lennartsson, Ola Spjuth, Ola Söderberg\",\"doi\":\"10.1038/s41467-025-62512-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The ability to analyze whether DNA contains lesions is essential in identifying mutagenic substances. Currently, the detection of single-stranded DNA breaks (SSBs) lacks precision. To address this limitation, we develop a method for sequence-templated erroneous end-labelling sequencing (STEEL-seq), which enables the mapping of SSBs. The method requires a highly error-prone DNA polymerase, so we engineer a chimeric DNA polymerase, Sloppymerase, capable of replicating DNA in the absence of one nucleotide. Following the omission of a specific nucleotide (e.g., dATP) from the reaction mixture, Sloppymerase introduces mismatches directly downstream of SSBs at positions where deoxyadenosine should occur. This mismatch pattern, coupled with the retention of sequence information flanking these sites, ensures that the identified hits are bona fide SSBs. STEEL-seq is compatible with a variety of sequencing technologies, as demonstrated using Sanger, Illumina, PacBio, and Nanopore systems. Using STEEL-seq, we determine the SSB/base pair frequency in the human genome to range between 0.7 and 3.8 × 10<sup>−6</sup> with an enrichment in active promoter regions.</p>\",\"PeriodicalId\":19066,\"journal\":{\"name\":\"Nature Communications\",\"volume\":\"19 1\",\"pages\":\"\"},\"PeriodicalIF\":15.7000,\"publicationDate\":\"2025-08-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature Communications\",\"FirstCategoryId\":\"103\",\"ListUrlMain\":\"https://doi.org/10.1038/s41467-025-62512-4\",\"RegionNum\":1,\"RegionCategory\":\"综合性期刊\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MULTIDISCIPLINARY SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Communications","FirstCategoryId":"103","ListUrlMain":"https://doi.org/10.1038/s41467-025-62512-4","RegionNum":1,"RegionCategory":"综合性期刊","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
Precise mapping of single-stranded DNA breaks by sequence-templated erroneous DNA polymerase end-labelling
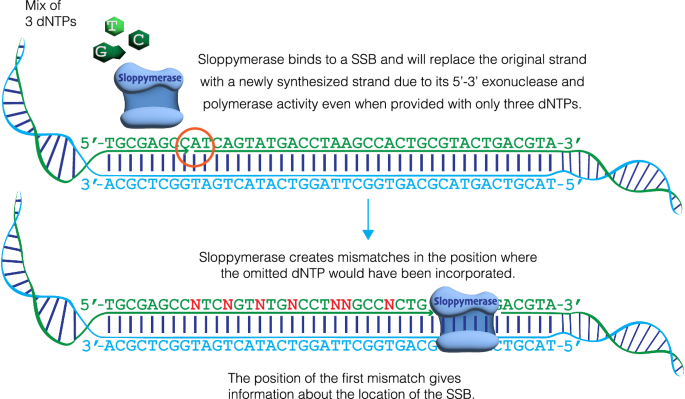
The ability to analyze whether DNA contains lesions is essential in identifying mutagenic substances. Currently, the detection of single-stranded DNA breaks (SSBs) lacks precision. To address this limitation, we develop a method for sequence-templated erroneous end-labelling sequencing (STEEL-seq), which enables the mapping of SSBs. The method requires a highly error-prone DNA polymerase, so we engineer a chimeric DNA polymerase, Sloppymerase, capable of replicating DNA in the absence of one nucleotide. Following the omission of a specific nucleotide (e.g., dATP) from the reaction mixture, Sloppymerase introduces mismatches directly downstream of SSBs at positions where deoxyadenosine should occur. This mismatch pattern, coupled with the retention of sequence information flanking these sites, ensures that the identified hits are bona fide SSBs. STEEL-seq is compatible with a variety of sequencing technologies, as demonstrated using Sanger, Illumina, PacBio, and Nanopore systems. Using STEEL-seq, we determine the SSB/base pair frequency in the human genome to range between 0.7 and 3.8 × 10−6 with an enrichment in active promoter regions.
期刊介绍:
Nature Communications, an open-access journal, publishes high-quality research spanning all areas of the natural sciences. Papers featured in the journal showcase significant advances relevant to specialists in each respective field. With a 2-year impact factor of 16.6 (2022) and a median time of 8 days from submission to the first editorial decision, Nature Communications is committed to rapid dissemination of research findings. As a multidisciplinary journal, it welcomes contributions from biological, health, physical, chemical, Earth, social, mathematical, applied, and engineering sciences, aiming to highlight important breakthroughs within each domain.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


