通过242个全基因组重排分析鼠疫耶尔森氏菌的进化
IF 29
1区 生物学
Q1 GENETICS & HEREDITY
引用次数: 0
摘要
鼠疫耶尔森氏菌是引起鼠疫的细菌,它有一个动态基因组,其高度保守的片段容易重排,影响基因功能和进化。然而,了解这些模式受到少数完整基因组和分析方法的限制。我们开发了一种双重验证策略来分析来自不同种群的242个鼠疫杆菌天然分离物的完整基因组。我们检测到459个重排,这增强了系统发育分辨率,并解决了第三次大流行的多灶性。重排主要由四个常见的插入序列介导,其中IS1661和IS100表现出最高的活性。rpsO-pnp操纵子的43个热点和趋同进化证明了这些重排是在强烈的正选择下进行的,它们的破坏和重连接改变了基因的表达和温度胁迫反应。我们还发现了人类无毒系统群中独特的结构变化,使3个基因失活,并重新排序了17个基因间区域,其中一些影响毒性相关基因。本研究为鼠疫杆菌的进化提供了新的视角,揭示了实验靶点,并建立了一种研究频繁重排微生物的方法。本文章由计算机程序翻译,如有差异,请以英文原文为准。
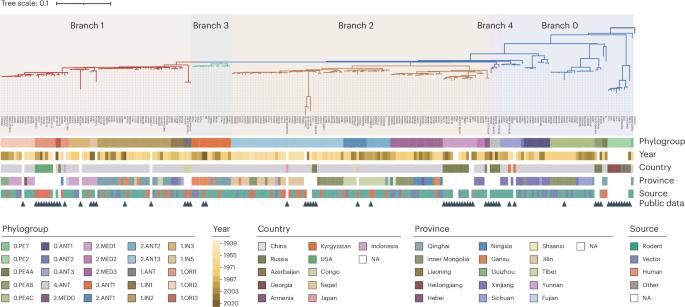

Insights into Yersinia pestis evolution through rearrangement analysis of 242 complete genomes
Yersinia pestis, the bacterium that causes the plague, has a dynamic genome with highly conserved fragments prone to rearrangement, influencing gene function and evolution. However, understanding these patterns is limited by few complete genomes and analytical methods. We developed a dual-validation strategy to analyze 242 complete genomes of Y. pestis natural isolates from diverse phylogroups. We detected 459 rearrangements, which enhanced phylogenetic resolution and resolved the third pandemic’s polytomy. Rearrangements are primarily mediated by four common insertion sequences, with IS1661 and IS100 showing the highest activity. These rearrangements are under strong positive selection, evidenced by 43 hotspots and convergent evolution in the rpsO-pnp operon, whose disruptions and reconnections altered gene expressions and temperature stress responses. We also identified unique structural alterations in human avirulent phylogroups, inactivating three genes and reordering 17 intergenic regions, some affecting virulence-related genes. This study provides a fresh perspective on Y. pestis evolution, revealing experimental targets and establishing a methodology for microbes with frequent rearrangements. Analysis of synteny blocks and genomic rearrangement patterns of 178 newly sequenced and 64 publicly available complete genomes of Yersinia pestis highlights the impact of key variations on genes involved in adaptation and virulence.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Nature genetics
生物-遗传学
CiteScore
43.00
自引率
2.60%
发文量
241
审稿时长
3 months
期刊介绍:
Nature Genetics publishes the very highest quality research in genetics. It encompasses genetic and functional genomic studies on human and plant traits and on other model organisms. Current emphasis is on the genetic basis for common and complex diseases and on the functional mechanism, architecture and evolution of gene networks, studied by experimental perturbation.
Integrative genetic topics comprise, but are not limited to:
-Genes in the pathology of human disease
-Molecular analysis of simple and complex genetic traits
-Cancer genetics
-Agricultural genomics
-Developmental genetics
-Regulatory variation in gene expression
-Strategies and technologies for extracting function from genomic data
-Pharmacological genomics
-Genome evolution
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


