{"title":"含异构体杂环醛的Criegee中间体(ch260)的温度和压力依赖动力学:理论研究。","authors":"Amit Debnath*, and , Balla Rajakumar, ","doi":"10.1021/acs.jpca.5c02887","DOIUrl":null,"url":null,"abstract":"<p >Temperature- and pressure-dependent kinetic study of the reactions of the Criegee intermediate (CH<sub>2</sub>OO) with 2-furaldehyde and 3-furaldehyde was performed by using computational methodologies. The geometry optimization and thermochemical parameters calculations were performed using B3LYP/6-311G+(2df,2p) theory, whereas the barrier heights in the potential energy diagram for the reaction pathways were calculated at the CCSD(T)-F12b/cc-pVTZ-F12//B3LYP/6-311 + G(2df,2p) level of theory. The room-temperature high-pressure-limit rate coefficients were calculated using canonical variational transition state theory in conjugation with small curvature tunneling (CVT/SCT) to be 7.20 × 10<sup>–12</sup> cm<sup>3</sup> molecule<sup>–1</sup> s<sup>–1</sup> and 3.04 × 10<sup>–12</sup> cm<sup>3</sup> molecule<sup>–1</sup> s<sup>–1</sup> for 2-furaldehyde and 3-furaldehyde, respectively. The room-temperature rate coefficients calculated from the MESMER were 2.62 × 10<sup>–12</sup> cm<sup>3</sup> molecule<sup>–1</sup> s<sup>–1</sup> and 1.59 × 10<sup>–12</sup> cm<sup>3</sup> molecule<sup>–1</sup> s<sup>–1</sup>, respectively. The predicted products of the title reactions were formic acid and formaldehyde, along with 2-furoic acid for the 2-furaldehyde reaction and 3-furoic acid for the 3-furaldehyde reaction. The large value of atmospheric lifetimes (>400 days) of the furaldehydes due to their reactions with CH<sub>2</sub>OO suggests that the title reactions do not have any significant effect on the net atmospheric furaldehyde concentrations.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"129 32","pages":"7470–7481"},"PeriodicalIF":2.8000,"publicationDate":"2025-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Temperature- and Pressure-Dependent Kinetics of the Criegee Intermediate (CH2OO) with Isomeric Heterocyclic Aldehyde: A Theoretical Study\",\"authors\":\"Amit Debnath*, and , Balla Rajakumar, \",\"doi\":\"10.1021/acs.jpca.5c02887\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Temperature- and pressure-dependent kinetic study of the reactions of the Criegee intermediate (CH<sub>2</sub>OO) with 2-furaldehyde and 3-furaldehyde was performed by using computational methodologies. The geometry optimization and thermochemical parameters calculations were performed using B3LYP/6-311G+(2df,2p) theory, whereas the barrier heights in the potential energy diagram for the reaction pathways were calculated at the CCSD(T)-F12b/cc-pVTZ-F12//B3LYP/6-311 + G(2df,2p) level of theory. The room-temperature high-pressure-limit rate coefficients were calculated using canonical variational transition state theory in conjugation with small curvature tunneling (CVT/SCT) to be 7.20 × 10<sup>–12</sup> cm<sup>3</sup> molecule<sup>–1</sup> s<sup>–1</sup> and 3.04 × 10<sup>–12</sup> cm<sup>3</sup> molecule<sup>–1</sup> s<sup>–1</sup> for 2-furaldehyde and 3-furaldehyde, respectively. The room-temperature rate coefficients calculated from the MESMER were 2.62 × 10<sup>–12</sup> cm<sup>3</sup> molecule<sup>–1</sup> s<sup>–1</sup> and 1.59 × 10<sup>–12</sup> cm<sup>3</sup> molecule<sup>–1</sup> s<sup>–1</sup>, respectively. The predicted products of the title reactions were formic acid and formaldehyde, along with 2-furoic acid for the 2-furaldehyde reaction and 3-furoic acid for the 3-furaldehyde reaction. The large value of atmospheric lifetimes (>400 days) of the furaldehydes due to their reactions with CH<sub>2</sub>OO suggests that the title reactions do not have any significant effect on the net atmospheric furaldehyde concentrations.</p>\",\"PeriodicalId\":59,\"journal\":{\"name\":\"The Journal of Physical Chemistry A\",\"volume\":\"129 32\",\"pages\":\"7470–7481\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2025-08-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry A\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpca.5c02887\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.5c02887","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Temperature- and Pressure-Dependent Kinetics of the Criegee Intermediate (CH2OO) with Isomeric Heterocyclic Aldehyde: A Theoretical Study
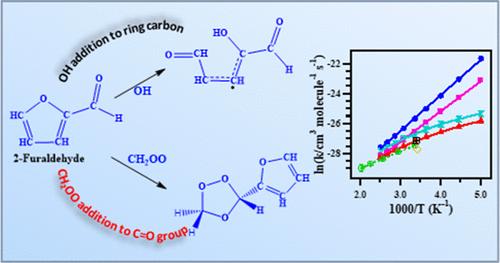
Temperature- and pressure-dependent kinetic study of the reactions of the Criegee intermediate (CH2OO) with 2-furaldehyde and 3-furaldehyde was performed by using computational methodologies. The geometry optimization and thermochemical parameters calculations were performed using B3LYP/6-311G+(2df,2p) theory, whereas the barrier heights in the potential energy diagram for the reaction pathways were calculated at the CCSD(T)-F12b/cc-pVTZ-F12//B3LYP/6-311 + G(2df,2p) level of theory. The room-temperature high-pressure-limit rate coefficients were calculated using canonical variational transition state theory in conjugation with small curvature tunneling (CVT/SCT) to be 7.20 × 10–12 cm3 molecule–1 s–1 and 3.04 × 10–12 cm3 molecule–1 s–1 for 2-furaldehyde and 3-furaldehyde, respectively. The room-temperature rate coefficients calculated from the MESMER were 2.62 × 10–12 cm3 molecule–1 s–1 and 1.59 × 10–12 cm3 molecule–1 s–1, respectively. The predicted products of the title reactions were formic acid and formaldehyde, along with 2-furoic acid for the 2-furaldehyde reaction and 3-furoic acid for the 3-furaldehyde reaction. The large value of atmospheric lifetimes (>400 days) of the furaldehydes due to their reactions with CH2OO suggests that the title reactions do not have any significant effect on the net atmospheric furaldehyde concentrations.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


