{"title":"维管起源的镶嵌现象","authors":"Olivia Boccara","doi":"10.1002/jvc2.70074","DOIUrl":null,"url":null,"abstract":"<p>Superficial vascular anomalies are mosaic disorders and are mostly sporadic. They result from activating post-zygotic mutations involving genes that belong to two main signalling pathways: the PIK3CA-AKT-mTOR pathway and the RAS-MAK-kinases pathway. More rarely, some conditions may be hereditary, and mosaic clinical manifestations result from the second hit mechanism; in those cases, the pathogenic variant leads to a loss-of-function; the capillary malformation-arteriovenous malformation syndrome (CM-AVM) is related to pathogenic variants in <i>RASA1</i> or <i>EPHB4</i>; the <i>PTEN</i> hamartoma and tumour syndrome (PHTS) results from <i>PTEN</i> pathogenic variants [<span>1</span>].</p><p>Superficial vascular anomalies are listed in the ISSVA classification [<span>2</span>]. Several guidelines for management of various types of vascular anomalies are already published by different teams, and it is not our aim herein to add a further opinion [<span>1-4</span>]. The overall management of cutaneous mosaic disorders described by Kinsler et al. feats perfectly well those of vascular origin [<span>5</span>]. Diagnosis relies first on clinical manifestations that can be enough to make it out. However, in some instances, further investigations may be necessary: imaging such as US Doppler, Magnetic resonance imaging and angiography, histopathology and genetics. Investigations will be specifically performed depending on the complete clinical assessment conclusions. Genetics is not always mandatory and frequently not enough to confirm a diagnosis. A single lesion may be related to different pathogenic variants. This is the case of venous malformations that can result from either <i>TEK</i> or <i>PIK3CA</i> mutations [<span>6, 7</span>]. As another example, soft tissue angiomatosis, which is characterized by specific clinical, imaging and histopathological features, will be defined as PHTS hamartoma of soft tissue (PHOST) in the context of PHTS, and Fibro-adipose vascular anomaly (FAVA) when isolated with an identified somatic <i>PIK3CA</i> pathogenic variant [<span>8</span>]. On the other hand, a single pathogenic variant may lead to different types of lesions or syndromes: <i>PIK3CA</i> will be responsible for isolated venous and lymphatic malformations, combined syndromes such as Congenital Lipomatous Overgrowth Vascular Epidermal skeletal anomalies syndrome (CLOVES) or megalencephaly-capillary malformation (M-CAP) [<span>9</span>]. Therefore, identified genetic variants always need to be confronted with clinical manifestations as well as imaging or histopathological features, if necessary, to reach an accurate diagnosis. Even more, some clinical presentations may be highly evocative of a specific syndrome, but the expected pathogenic variant fails to be identified in some instances. In the CM-AVM syndrome characterized by numerous small round pale pink capillary malformations with a pale halo, central nervous system screening may be considered to search for brain or spine arteriovenous malformation, even if the expected mutation is not identified [<span>10</span>]. This means that the diagnosis of CM-AVM syndrome relies more on the clinical manifestations than the genetic analysis.</p><p>Such as for CM-AVM syndrome, we strongly would advise to use as much as possible a more descriptive terminology, which should be as close as possible to pathophysiological mechanisms to simplify and unify the terminology and to avoid misunderstanding and confusion. Indeed in the past, diseases were frequently designated by surnames or other terms that could be confusing. As an example, the so-called “Klippel-Trenaunay syndrome” should be better named by the different involved vascular and tissular components: capillaro-veno-lymphatic malformation with overgrowth. This should avoid using “Klippel-Trenaunay syndrome” for any kind of limb overgrowth with overlying capillary malformation. Furthermore, such a descriptive terminology would also allow naming correctly overlapping or incomplete presentations, that do not fit exactly well-delineated entities. As an example, some infiltrative lymphatic malformations, without venous component, may be complicated by pain and mild chronic coagulopathy, but without kaposiform morphology on histological analysis; they cannot be considered as a simple lymphatic malformation, but they cannot be named kaposiform hemangioendothelioma or tufted angioma or kaposiform lymphangiomatosis. In parallel, the increasing knowledge about genetic basis of vascular anomalies shows that mutations lead to various lesions that may share similar features, and actually comprise a continuous spectrum of diseases. The <i>PIK3CA</i>-related disorders illustrate very well this concept [<span>9</span>]; however, it is also true for the <i>RAS-MAP</i>-Kinases pathway, even if probably less reported (Figure 1). Capillary malformations related to <i>GNAQ</i> or <i>GNA11</i> variants may be associated with phlebectasia, which are sometimes seen on residual lesions of congenital hemangiomas [<span>11, 12</span>]. <i>GNAQ</i> related capillary malformations may be hypertrophic and characterized by high-flow on imaging, such as in AVM [<span>13</span>], but also congenital hemangioma and kaposiform henmangioendothelioma and tufted angioma; those vascular tumours comprise a lymphatic component sometimes observed in association with arteriovenous malformation, and a more or less severe coagulopathy which is encountered in atypical lymphatic anomalies (generalized lymphatic anomaly, kaposiform lymphatic anomaly). Indeed, such as in the PIK3CA-AKT-MTOR spectrum, the vascular malformations from the RAS-MAP-Kinases pathway share several features that result in the lesional continuum.</p><p>Based on increasing knowledge in vascular malformations, mainly the development of genetic testing, the number of therapeutic options is increasing, comprising targeted therapies [<span>14, 15</span>]. Several fundamentals need to be kept in mind: Superficial vascular lesions are benign lesions. No curative treatment is available for any of them, except for very small lesions for which surgical removal can be easily performed. This means that vascular anomalies are chronic, long-lasting disorders and long-term clinical follow-up is needed. Treatment strategy is adapted on a case-by-case basis, targeting the patient's specific symptoms that may evolve overtime.</p><p>The author has drafted the manuscript and critically reviewed its content autonomously.</p><p>The author has nothing to report.</p><p>The author declares no conflicts of interest.</p>","PeriodicalId":94325,"journal":{"name":"JEADV clinical practice","volume":"4 3","pages":"697-699"},"PeriodicalIF":0.5000,"publicationDate":"2025-08-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jvc2.70074","citationCount":"0","resultStr":"{\"title\":\"Mosaicism of Vascular Origin\",\"authors\":\"Olivia Boccara\",\"doi\":\"10.1002/jvc2.70074\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Superficial vascular anomalies are mosaic disorders and are mostly sporadic. They result from activating post-zygotic mutations involving genes that belong to two main signalling pathways: the PIK3CA-AKT-mTOR pathway and the RAS-MAK-kinases pathway. More rarely, some conditions may be hereditary, and mosaic clinical manifestations result from the second hit mechanism; in those cases, the pathogenic variant leads to a loss-of-function; the capillary malformation-arteriovenous malformation syndrome (CM-AVM) is related to pathogenic variants in <i>RASA1</i> or <i>EPHB4</i>; the <i>PTEN</i> hamartoma and tumour syndrome (PHTS) results from <i>PTEN</i> pathogenic variants [<span>1</span>].</p><p>Superficial vascular anomalies are listed in the ISSVA classification [<span>2</span>]. Several guidelines for management of various types of vascular anomalies are already published by different teams, and it is not our aim herein to add a further opinion [<span>1-4</span>]. The overall management of cutaneous mosaic disorders described by Kinsler et al. feats perfectly well those of vascular origin [<span>5</span>]. Diagnosis relies first on clinical manifestations that can be enough to make it out. However, in some instances, further investigations may be necessary: imaging such as US Doppler, Magnetic resonance imaging and angiography, histopathology and genetics. Investigations will be specifically performed depending on the complete clinical assessment conclusions. Genetics is not always mandatory and frequently not enough to confirm a diagnosis. A single lesion may be related to different pathogenic variants. This is the case of venous malformations that can result from either <i>TEK</i> or <i>PIK3CA</i> mutations [<span>6, 7</span>]. As another example, soft tissue angiomatosis, which is characterized by specific clinical, imaging and histopathological features, will be defined as PHTS hamartoma of soft tissue (PHOST) in the context of PHTS, and Fibro-adipose vascular anomaly (FAVA) when isolated with an identified somatic <i>PIK3CA</i> pathogenic variant [<span>8</span>]. On the other hand, a single pathogenic variant may lead to different types of lesions or syndromes: <i>PIK3CA</i> will be responsible for isolated venous and lymphatic malformations, combined syndromes such as Congenital Lipomatous Overgrowth Vascular Epidermal skeletal anomalies syndrome (CLOVES) or megalencephaly-capillary malformation (M-CAP) [<span>9</span>]. Therefore, identified genetic variants always need to be confronted with clinical manifestations as well as imaging or histopathological features, if necessary, to reach an accurate diagnosis. Even more, some clinical presentations may be highly evocative of a specific syndrome, but the expected pathogenic variant fails to be identified in some instances. In the CM-AVM syndrome characterized by numerous small round pale pink capillary malformations with a pale halo, central nervous system screening may be considered to search for brain or spine arteriovenous malformation, even if the expected mutation is not identified [<span>10</span>]. This means that the diagnosis of CM-AVM syndrome relies more on the clinical manifestations than the genetic analysis.</p><p>Such as for CM-AVM syndrome, we strongly would advise to use as much as possible a more descriptive terminology, which should be as close as possible to pathophysiological mechanisms to simplify and unify the terminology and to avoid misunderstanding and confusion. Indeed in the past, diseases were frequently designated by surnames or other terms that could be confusing. As an example, the so-called “Klippel-Trenaunay syndrome” should be better named by the different involved vascular and tissular components: capillaro-veno-lymphatic malformation with overgrowth. This should avoid using “Klippel-Trenaunay syndrome” for any kind of limb overgrowth with overlying capillary malformation. Furthermore, such a descriptive terminology would also allow naming correctly overlapping or incomplete presentations, that do not fit exactly well-delineated entities. As an example, some infiltrative lymphatic malformations, without venous component, may be complicated by pain and mild chronic coagulopathy, but without kaposiform morphology on histological analysis; they cannot be considered as a simple lymphatic malformation, but they cannot be named kaposiform hemangioendothelioma or tufted angioma or kaposiform lymphangiomatosis. In parallel, the increasing knowledge about genetic basis of vascular anomalies shows that mutations lead to various lesions that may share similar features, and actually comprise a continuous spectrum of diseases. The <i>PIK3CA</i>-related disorders illustrate very well this concept [<span>9</span>]; however, it is also true for the <i>RAS-MAP</i>-Kinases pathway, even if probably less reported (Figure 1). Capillary malformations related to <i>GNAQ</i> or <i>GNA11</i> variants may be associated with phlebectasia, which are sometimes seen on residual lesions of congenital hemangiomas [<span>11, 12</span>]. <i>GNAQ</i> related capillary malformations may be hypertrophic and characterized by high-flow on imaging, such as in AVM [<span>13</span>], but also congenital hemangioma and kaposiform henmangioendothelioma and tufted angioma; those vascular tumours comprise a lymphatic component sometimes observed in association with arteriovenous malformation, and a more or less severe coagulopathy which is encountered in atypical lymphatic anomalies (generalized lymphatic anomaly, kaposiform lymphatic anomaly). Indeed, such as in the PIK3CA-AKT-MTOR spectrum, the vascular malformations from the RAS-MAP-Kinases pathway share several features that result in the lesional continuum.</p><p>Based on increasing knowledge in vascular malformations, mainly the development of genetic testing, the number of therapeutic options is increasing, comprising targeted therapies [<span>14, 15</span>]. Several fundamentals need to be kept in mind: Superficial vascular lesions are benign lesions. No curative treatment is available for any of them, except for very small lesions for which surgical removal can be easily performed. This means that vascular anomalies are chronic, long-lasting disorders and long-term clinical follow-up is needed. Treatment strategy is adapted on a case-by-case basis, targeting the patient's specific symptoms that may evolve overtime.</p><p>The author has drafted the manuscript and critically reviewed its content autonomously.</p><p>The author has nothing to report.</p><p>The author declares no conflicts of interest.</p>\",\"PeriodicalId\":94325,\"journal\":{\"name\":\"JEADV clinical practice\",\"volume\":\"4 3\",\"pages\":\"697-699\"},\"PeriodicalIF\":0.5000,\"publicationDate\":\"2025-08-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jvc2.70074\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"JEADV clinical practice\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jvc2.70074\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"JEADV clinical practice","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jvc2.70074","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Superficial vascular anomalies are mosaic disorders and are mostly sporadic. They result from activating post-zygotic mutations involving genes that belong to two main signalling pathways: the PIK3CA-AKT-mTOR pathway and the RAS-MAK-kinases pathway. More rarely, some conditions may be hereditary, and mosaic clinical manifestations result from the second hit mechanism; in those cases, the pathogenic variant leads to a loss-of-function; the capillary malformation-arteriovenous malformation syndrome (CM-AVM) is related to pathogenic variants in RASA1 or EPHB4; the PTEN hamartoma and tumour syndrome (PHTS) results from PTEN pathogenic variants [1].
Superficial vascular anomalies are listed in the ISSVA classification [2]. Several guidelines for management of various types of vascular anomalies are already published by different teams, and it is not our aim herein to add a further opinion [1-4]. The overall management of cutaneous mosaic disorders described by Kinsler et al. feats perfectly well those of vascular origin [5]. Diagnosis relies first on clinical manifestations that can be enough to make it out. However, in some instances, further investigations may be necessary: imaging such as US Doppler, Magnetic resonance imaging and angiography, histopathology and genetics. Investigations will be specifically performed depending on the complete clinical assessment conclusions. Genetics is not always mandatory and frequently not enough to confirm a diagnosis. A single lesion may be related to different pathogenic variants. This is the case of venous malformations that can result from either TEK or PIK3CA mutations [6, 7]. As another example, soft tissue angiomatosis, which is characterized by specific clinical, imaging and histopathological features, will be defined as PHTS hamartoma of soft tissue (PHOST) in the context of PHTS, and Fibro-adipose vascular anomaly (FAVA) when isolated with an identified somatic PIK3CA pathogenic variant [8]. On the other hand, a single pathogenic variant may lead to different types of lesions or syndromes: PIK3CA will be responsible for isolated venous and lymphatic malformations, combined syndromes such as Congenital Lipomatous Overgrowth Vascular Epidermal skeletal anomalies syndrome (CLOVES) or megalencephaly-capillary malformation (M-CAP) [9]. Therefore, identified genetic variants always need to be confronted with clinical manifestations as well as imaging or histopathological features, if necessary, to reach an accurate diagnosis. Even more, some clinical presentations may be highly evocative of a specific syndrome, but the expected pathogenic variant fails to be identified in some instances. In the CM-AVM syndrome characterized by numerous small round pale pink capillary malformations with a pale halo, central nervous system screening may be considered to search for brain or spine arteriovenous malformation, even if the expected mutation is not identified [10]. This means that the diagnosis of CM-AVM syndrome relies more on the clinical manifestations than the genetic analysis.
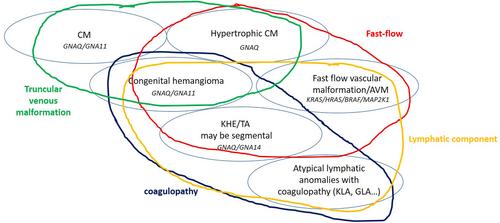
Such as for CM-AVM syndrome, we strongly would advise to use as much as possible a more descriptive terminology, which should be as close as possible to pathophysiological mechanisms to simplify and unify the terminology and to avoid misunderstanding and confusion. Indeed in the past, diseases were frequently designated by surnames or other terms that could be confusing. As an example, the so-called “Klippel-Trenaunay syndrome” should be better named by the different involved vascular and tissular components: capillaro-veno-lymphatic malformation with overgrowth. This should avoid using “Klippel-Trenaunay syndrome” for any kind of limb overgrowth with overlying capillary malformation. Furthermore, such a descriptive terminology would also allow naming correctly overlapping or incomplete presentations, that do not fit exactly well-delineated entities. As an example, some infiltrative lymphatic malformations, without venous component, may be complicated by pain and mild chronic coagulopathy, but without kaposiform morphology on histological analysis; they cannot be considered as a simple lymphatic malformation, but they cannot be named kaposiform hemangioendothelioma or tufted angioma or kaposiform lymphangiomatosis. In parallel, the increasing knowledge about genetic basis of vascular anomalies shows that mutations lead to various lesions that may share similar features, and actually comprise a continuous spectrum of diseases. The PIK3CA-related disorders illustrate very well this concept [9]; however, it is also true for the RAS-MAP-Kinases pathway, even if probably less reported (Figure 1). Capillary malformations related to GNAQ or GNA11 variants may be associated with phlebectasia, which are sometimes seen on residual lesions of congenital hemangiomas [11, 12]. GNAQ related capillary malformations may be hypertrophic and characterized by high-flow on imaging, such as in AVM [13], but also congenital hemangioma and kaposiform henmangioendothelioma and tufted angioma; those vascular tumours comprise a lymphatic component sometimes observed in association with arteriovenous malformation, and a more or less severe coagulopathy which is encountered in atypical lymphatic anomalies (generalized lymphatic anomaly, kaposiform lymphatic anomaly). Indeed, such as in the PIK3CA-AKT-MTOR spectrum, the vascular malformations from the RAS-MAP-Kinases pathway share several features that result in the lesional continuum.
Based on increasing knowledge in vascular malformations, mainly the development of genetic testing, the number of therapeutic options is increasing, comprising targeted therapies [14, 15]. Several fundamentals need to be kept in mind: Superficial vascular lesions are benign lesions. No curative treatment is available for any of them, except for very small lesions for which surgical removal can be easily performed. This means that vascular anomalies are chronic, long-lasting disorders and long-term clinical follow-up is needed. Treatment strategy is adapted on a case-by-case basis, targeting the patient's specific symptoms that may evolve overtime.
The author has drafted the manuscript and critically reviewed its content autonomously.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


