水溶性纳米笼内主客体电荷转移介导的光氧化还原催化。
摘要
可见光介导的光氧化还原催化,继Richard Kellogg和Shunichi Fukuzumi对光敏化反应的早期研究之后,于2008年由David MacMillan复兴,已被用作在温和反应条件下进行复杂有机合成的有效策略。在选择性光氧化还原转化过程中使用可见光的主要动机是尽量减少高能和化学破坏性紫外线光子的使用,从而模拟自然光合作用。具有大量可见光吸收截面和长三重态激发态寿命的过渡金属基光敏剂已经成功地用于有效地驱动有机溶剂中光介导的化学转化。另一种执行可见光介导光催化的方法是利用建立稳定的供体-受体电荷转移(CT)相互作用的思想,正如Melchiorre及其同事在2013年所证明的那样,他们利用基于CT的催化在大量有机溶剂中进行立体选择性C-C偶联反应。一般来说,大多数当代光催化有机转化在散装溶剂中通过扩散限制过程进行,通常使用三重态。然而,如果在扩散驱动的反应性碰撞中,激发能转移(EnT)和/或电子转移(ET)在时间上没有最优的时间尺度,光子能量利用率就会很低,因此通常使用高功率光源。因此,能够在水中进行超快光化学催化转化的工程预组织供体-受体界面将在模仿天然酶的同时彻底改变光氧化还原范式。自2014年以来,我们一直致力于在水溶性超分子纳米笼中开发可见光驱动的主-客体CT状态介导的光氧化还原催化。概念框架依赖于使用缺电子、阳离子的超分子腔来建立CT与氧化还原互补、富电子和不溶于水的有机客体分子的相互作用。由于自组装在阳离子Pd6L412+纳米腔内的客体预组织中起主导作用,我们成功地展示了可见光诱导的O-H, N-H和C-H键光激活以及在室温下使用低功率可见光源的光催化C-C偶联反应的罕见例子。基于纳米笼的催化循环的设计原则依赖于互补的供体-受体氧化还原对、激励态主客体电荷转移动力学、约束几何以及水辅助质子耦合电子转移(PCET)反应的战略性使用。在这里,我们系统地叙述了我们的概念框架,强调光激发单线态CT态的使用,以便通过水溶性纳米笼内的预组织发现一种新的键激活范式。我们描述了使用宽带飞秒瞬态吸收(fs-TA)光谱来记录纳米笼周围水的超快溶剂化动力学,同时观察O-H, N-H和C-H键断裂步骤,通过耦合电子转移和质子转移步骤(尽管是暂时分离的)来实现。在驱动sp3-、sp2-和sp-杂化C-H键选择性氧化反应的背景下,确定光生成的笼状自由基的瞬态吸收特征及其各自的寿命具有重要意义;重点介绍了室温下甲苯到苯甲醛的首次选择性转化。我们进一步表明,我们的主客体ct介导的光化学方法是足够普遍的,因为它允许sp3-C-H键在全有机阳离子超分子腔内功能化。最后,我们深入研究了调节这种纳米笼的限制几何,电子和体积以完全改变产品分布的必要性,这将是所有合成有机化学家的有用处理方法。
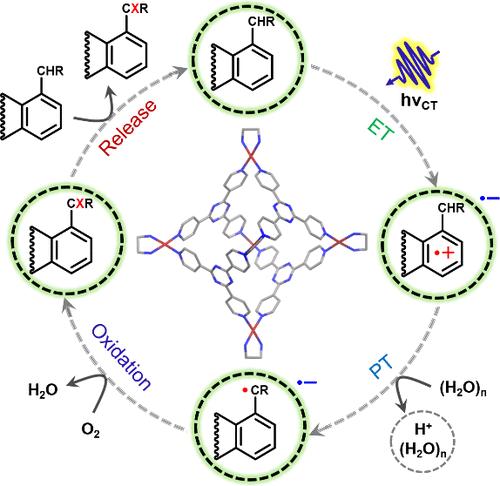
Visible-light-mediated photoredox catalysis, rejuvenated by David MacMillan in 2008 following the early work by Richard Kellogg and Shunichi Fukuzumi on photosensitized reactions, has been used as an efficient strategy to carry out complex organic synthesis under mild reaction conditions. The main motivation for using visible-light during selective photoredox transformations is to minimize the use of high-energy and chemically disruptive UV-photons, thereby mimicking natural photosynthesis. Transition metal-based photosensitizers with substantial visible-light absorption cross sections and long triplet excited-state lifetimes have been successfully used to efficiently drive such light-mediated chemical transformations in organic solvents. An alternate approach for executing visible-light-mediated photocatalysis uses the idea of establishing stable donor–acceptor charge-transfer (CT) interactions, as demonstrated in 2013 by Melchiorre and co-workers, who utilized CT-based catalysis to carry out stereoselective C–C coupling reactions in bulk organic solvents. In general, most contemporary photocatalytic organic transformations in bulk solvents proceed via diffusion-limited processes, typically using triplet states. However, if the excitation energy transfer (EnT) and/or electron transfer (ET) are not temporally ordered with optimal timescales for reactive collisions driven by diffusion, photon-energy utilization will be low, due to which high-power light sources are usually applied. Therefore, engineering preorganized donor–acceptor interfaces capable of ultrafast photochemistry for catalytic transformation in water would revolutionize the photoredox paradigm while mimicking natural enzymes.
This Account focuses on our efforts since 2014 to develop visible-light-driven host–guest CT state-mediated photoredox catalysis within water-soluble supramolecular nanocages. The conceptual framework relies on using an electron-deficient, cationic supramolecular cavity to establish CT interactions with redox-complementary, electron-rich, and water-insoluble organic guest molecules. With self-assembly playing a dominant role in guest preorganization inside cationic Pd6L412+ nanocavity, we successfully demonstrated visible-light-induced O–H, N–H, and C–H bond photoactivation along with rare examples of photocatalytic C–C coupling reactions at room temperature using low-power visible-light sources. The design principles for nanocage-based catalytic cycles have relied on the strategic use of complementary donor–acceptor redox pairs, excited-state host–guest charge-transfer dynamics, confinement geometry, and the water-assisted proton-coupled electron transfer (PCET) reaction. Here, we systematically recount our conceptual framework, which emphasizes the usage of photoexcited singlet CT states in order to discover a novel bond-activation paradigm via preorganization inside water-soluble nanocages. We describe the use of broadband femtosecond transient absorption (fs-TA) spectroscopy to clock ultrafast solvation dynamics of water around the nanocage along with watching O–H, N–H, and C–H bond-breaking steps, enabled by coupling the electron transfer and proton transfer steps which are albeit temporally segregated. The significance of identifying transient absorption signatures of the photogenerated cage-confined radical species and their respective lifetimes is put in the context of driving selective oxidation reactions at sp3-, sp2-, and sp-hybridized C–H bonds; highlighting the first selective toluene-to-benzaldehyde transformation at room temperature. We further show that our methodology of host–guest CT-mediated photochemistry is general enough since it allows sp3-C–H bond functionalization inside an all-organic cationic supramolecular cavity. Finally, we delve into the necessity of modulating the confining geometry, electronics, and volume of such nanocages to completely alter product distributions, which will be a useful handle for all synthetic organic chemists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


