胶原IV在Gould综合征和Alport综合征
IF 39.8
1区 医学
Q1 UROLOGY & NEPHROLOGY
引用次数: 0
摘要
IV型胶原是哺乳动物中由6个基因编码的基膜成分(COL4Α1-COL4A6)。由这些基因编码的α-链组装成三种已知的异源三聚体-胶原α1α1α2(IV), α3α4α5(IV)和α5α5α6(IV) -提供结构并作为多功能信号传导平台。胶原蛋白的祖先超家族成员胶原α -1(IV)链(COL4Α1)和胶原α -2(IV)链(COL4Α2)存在于整个动物王国中,存在于所有发育中和最成熟的哺乳动物组织中。与这种广泛分布相一致的是,COL4A1和COL4A2的变异导致一种称为古尔德综合征(Gould syndrome, GS)的先天性多系统疾病,其特征是大脑、眼部、肌肉和肾脏缺陷。主要的临床后果涉及脑血管系统(脑孔畸形、小血管疾病、脑白质病和脑出血)。然而,完整的临床谱,包括受影响的器官和获得性表型,如血管性痴呆,仍在定义中。相比之下,COL4A3、COL4A4或COL4A5的变异会导致阿尔波特综合征(AS),这是一种影响肾脏、耳朵和眼睛的严重程度不一的疾病。AS肾病常发展为血尿、蛋白尿、肾功能损害和肾衰竭。听觉特征包括感音神经性听力损失,而眼部特征包括角膜营养不良、晶状体、斑点视网膜病变和黄斑病变。虽然GS和AS几乎没有临床相似性,但基因和蛋白质的高度保守性表明潜在病理生理的共同因素。改变血液动力学的传统疗法延长了AS患者发生肾衰竭的时间。然而,目前还没有根治性或基于机制的干预措施。基因编辑方法有望治疗这两种疾病。本文章由计算机程序翻译,如有差异,请以英文原文为准。
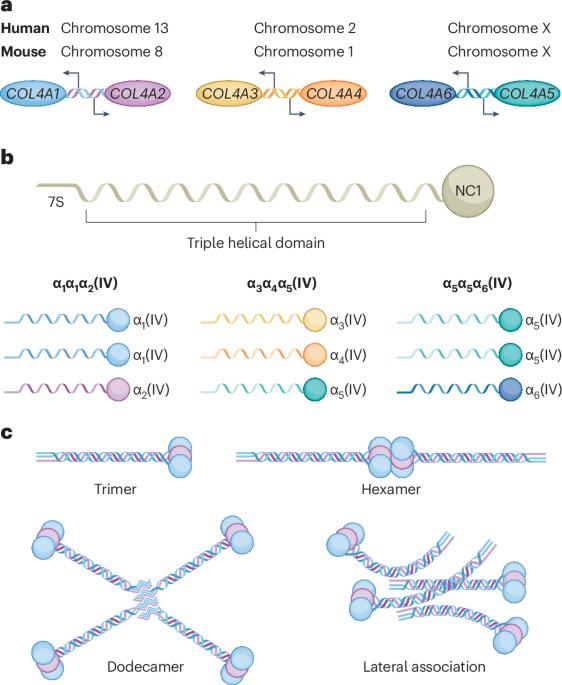
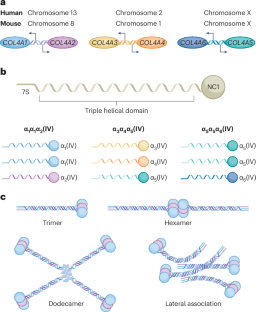
Collagen IV in Gould syndrome and Alport syndrome
Collagen IV is a basement membrane component that is encoded by six genes in mammals (COL4Α1–COL4A6). The α-chains encoded by these genes assemble into three known heterotrimers — collagen α1α1α2(IV), α3α4α5(IV) and α5α5α6(IV) — that provide structure and act as multifunctional signalling platforms. The ancestral collagen superfamily members collagen alpha-1(IV) chain (COL4Α1) and collagen alpha-2(IV) chain (COL4Α2) are present throughout the animal kingdom and in all developing and most mature mammalian tissues. Consistent with this broad distribution, variants in COL4A1 and COL4A2 cause a congenital multisystem disorder called Gould syndrome (GS), which is characterized by cerebral, ocular, muscular and kidney defects. The main clinical consequences involve the cerebral vasculature (porencephaly, small-vessel disease, leukoencephalopathy and intracerebral haemorrhage). However, the full clinical spectrum, including the organs affected and acquired phenotypes such as vascular dementia, is still being defined. By contrast, variants in COL4A3, COL4A4 or COL4A5 cause Alport syndrome (AS), a disorder of variable severity that affects the kidney, ear and eye. AS nephropathies often progress from haematuria to proteinuria, renal impairment and kidney failure. The auditory features include sensorineural hearing loss, whereas the ocular features comprise corneal dystrophy, lenticonus, dot-and-fleck retinopathy and maculopathy. Although GS and AS have little clinical resemblance, the high conservation of the genes and proteins suggests common elements of underlying pathophysiology. Conventional therapies that modify haemodynamics have lengthened the time to kidney failure for patients living with AS. However, no curative or mechanism-based interventions exist for GS. Gene-editing approaches hold promise for both disorders. In this Review, the authors focus on the role of collagen IV in Gould syndrome and Alport syndrome. They discuss the molecular and phenotypic similarities and differences between these syndromes, as well as potential targeted therapeutic strategies.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Nature Reviews Nephrology
医学-泌尿学与肾脏学
CiteScore
39.00
自引率
1.20%
发文量
127
审稿时长
6-12 weeks
期刊介绍:
Nature Reviews Nephrology aims to be the premier source of reviews and commentaries for the scientific communities it serves.
It strives to publish authoritative, accessible articles.
Articles are enhanced with clearly understandable figures, tables, and other display items.
Nature Reviews Nephrology publishes Research Highlights, News & Views, Comments, Reviews, Perspectives, and Consensus Statements.
The content is relevant to nephrologists and basic science researchers.
The broad scope of the journal ensures that the work reaches the widest possible audience.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


