María González-Sánchez, María Jesús Ramírez-Expósito, José Manuel Martínez-Martos
{"title":"婴儿神经轴突营养不良:pla2g6相关神经变性的分子机制和发病机制。","authors":"María González-Sánchez, María Jesús Ramírez-Expósito, José Manuel Martínez-Martos","doi":"10.3934/Neuroscience.2025011","DOIUrl":null,"url":null,"abstract":"<p><p>Infantile neuroaxonal dystrophy (INAD), also known as <i>PLA2G6</i>-associated neurodegeneration (PLAN), is a rare, early-onset, autosomal recessively inherited neurodegenerative disease belonging to the group of neurodegenerations with brain iron accumulation (NBIA). The main cause of this disease is bi-allelic mutations in the <i>PLA2G6</i> gene, which codes for the enzyme phospholipase A2 type VI. Clinically, it manifests with progressive neurodevelopmental impairment, psychomotor regression, movement disorders, and pyramidal signs. Initially described in the 1950s, the classical form presents in the first two years of life, although later-onset variants are recognized. At the neuropathological level, INAD is characterized by the presence of neuroaxonal spheroids, which are dilations of degenerated axons, located mainly in the white matter, basal ganglia, and cerebellum. INAD is considered a rare or ultra-rare disease, with an estimated prevalence of approximately 1 per million individuals. Diagnosis requires a comprehensive evaluation combining clinical with neuroimaging studies, mainly magnetic resonance imaging (MRI), and genetic analysis. MRI may reveal early cerebellar atrophy and a low-intensity signal in the globus pallidus on iron-sensitive sequences, indicative of iron accumulation. Currently, there is no curative treatment for INAD, so management focuses on providing palliative care and symptom control using a multidisciplinary approach. However, various therapeutic strategies are being investigated, including gene therapy to correct the genetic defect, as well as approaches to modulate pathological pathways such as lipid peroxidation and iron accumulation.</p>","PeriodicalId":7732,"journal":{"name":"AIMS Neuroscience","volume":"12 2","pages":"180-202"},"PeriodicalIF":2.7000,"publicationDate":"2025-05-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12287646/pdf/","citationCount":"0","resultStr":"{\"title\":\"Infantile neuroaxonal dystrophy: Molecular mechanisms and pathogenesis of PLA2G6-associated neurodegeneration.\",\"authors\":\"María González-Sánchez, María Jesús Ramírez-Expósito, José Manuel Martínez-Martos\",\"doi\":\"10.3934/Neuroscience.2025011\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Infantile neuroaxonal dystrophy (INAD), also known as <i>PLA2G6</i>-associated neurodegeneration (PLAN), is a rare, early-onset, autosomal recessively inherited neurodegenerative disease belonging to the group of neurodegenerations with brain iron accumulation (NBIA). The main cause of this disease is bi-allelic mutations in the <i>PLA2G6</i> gene, which codes for the enzyme phospholipase A2 type VI. Clinically, it manifests with progressive neurodevelopmental impairment, psychomotor regression, movement disorders, and pyramidal signs. Initially described in the 1950s, the classical form presents in the first two years of life, although later-onset variants are recognized. At the neuropathological level, INAD is characterized by the presence of neuroaxonal spheroids, which are dilations of degenerated axons, located mainly in the white matter, basal ganglia, and cerebellum. INAD is considered a rare or ultra-rare disease, with an estimated prevalence of approximately 1 per million individuals. Diagnosis requires a comprehensive evaluation combining clinical with neuroimaging studies, mainly magnetic resonance imaging (MRI), and genetic analysis. MRI may reveal early cerebellar atrophy and a low-intensity signal in the globus pallidus on iron-sensitive sequences, indicative of iron accumulation. Currently, there is no curative treatment for INAD, so management focuses on providing palliative care and symptom control using a multidisciplinary approach. However, various therapeutic strategies are being investigated, including gene therapy to correct the genetic defect, as well as approaches to modulate pathological pathways such as lipid peroxidation and iron accumulation.</p>\",\"PeriodicalId\":7732,\"journal\":{\"name\":\"AIMS Neuroscience\",\"volume\":\"12 2\",\"pages\":\"180-202\"},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2025-05-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12287646/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"AIMS Neuroscience\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3934/Neuroscience.2025011\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"NEUROSCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"AIMS Neuroscience","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3934/Neuroscience.2025011","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
Infantile neuroaxonal dystrophy: Molecular mechanisms and pathogenesis of PLA2G6-associated neurodegeneration.
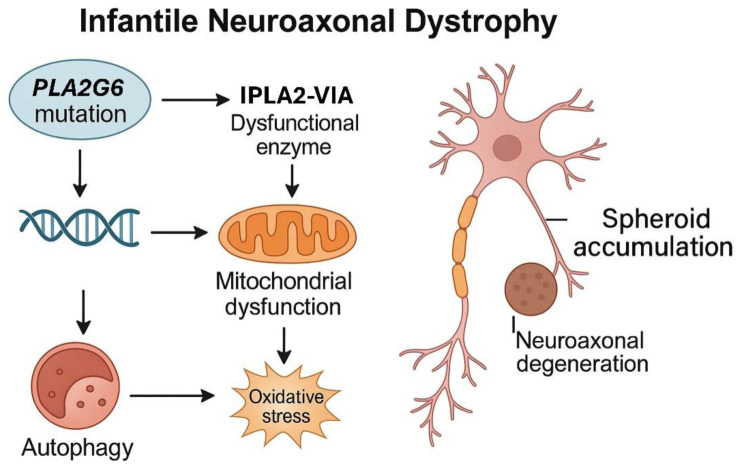
Infantile neuroaxonal dystrophy (INAD), also known as PLA2G6-associated neurodegeneration (PLAN), is a rare, early-onset, autosomal recessively inherited neurodegenerative disease belonging to the group of neurodegenerations with brain iron accumulation (NBIA). The main cause of this disease is bi-allelic mutations in the PLA2G6 gene, which codes for the enzyme phospholipase A2 type VI. Clinically, it manifests with progressive neurodevelopmental impairment, psychomotor regression, movement disorders, and pyramidal signs. Initially described in the 1950s, the classical form presents in the first two years of life, although later-onset variants are recognized. At the neuropathological level, INAD is characterized by the presence of neuroaxonal spheroids, which are dilations of degenerated axons, located mainly in the white matter, basal ganglia, and cerebellum. INAD is considered a rare or ultra-rare disease, with an estimated prevalence of approximately 1 per million individuals. Diagnosis requires a comprehensive evaluation combining clinical with neuroimaging studies, mainly magnetic resonance imaging (MRI), and genetic analysis. MRI may reveal early cerebellar atrophy and a low-intensity signal in the globus pallidus on iron-sensitive sequences, indicative of iron accumulation. Currently, there is no curative treatment for INAD, so management focuses on providing palliative care and symptom control using a multidisciplinary approach. However, various therapeutic strategies are being investigated, including gene therapy to correct the genetic defect, as well as approaches to modulate pathological pathways such as lipid peroxidation and iron accumulation.
期刊介绍:
AIMS Neuroscience is an international Open Access journal devoted to publishing peer-reviewed, high quality, original papers from all areas in the field of neuroscience. The primary focus is to provide a forum in which to expedite the speed with which theoretical neuroscience progresses toward generating testable hypotheses. In the presence of current and developing technology that offers unprecedented access to functions of the nervous system at all levels, the journal is designed to serve the role of providing the widest variety of the best theoretical views leading to suggested studies. Single blind peer review is provided for all articles and commentaries.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


