Shengyu Zhang, Donghui Huo, Robert I. Horne, Yumeng Qi, Sebastian Pujalte Ojeda, Aixia Yan, Michele Vendruscolo
{"title":"基于顺序的变压器虚拟筛选","authors":"Shengyu Zhang, Donghui Huo, Robert I. Horne, Yumeng Qi, Sebastian Pujalte Ojeda, Aixia Yan, Michele Vendruscolo","doi":"10.1038/s41467-025-61833-8","DOIUrl":null,"url":null,"abstract":"<p>Protein-ligand interactions play central roles in myriad biological processes and are of key importance in drug design. Deep learning approaches are becoming cost-effective alternatives to high-throughput experimental methods for ligand identification. Here, to predict the binding affinity between proteins and small molecules, we introduce Ligand-Transformer, a deep learning method based on the transformer architecture. Ligand-Transformer implements a sequence-based approach, where the inputs are the amino acid sequence of the target protein and the topology of the small molecule to enable the prediction of the conformational space explored by the complex between the two. We apply Ligand-Transformer to screen and validate experimentally inhibitors targeting the mutant EGFR<sup>LTC</sup> kinase, identifying compounds with low nanomolar potency. We then use this approach to predict the conformational population shifts induced by known ABL kinase inhibitors, showing that sequence-based predictions enable the characterisation of the population shift upon binding. Overall, our results illustrate the potential of Ligand-Transformer to accurately predict the interactions of small molecules with proteins, including the binding affinity and the changes in the free energy landscapes upon binding, thus uncovering molecular mechanisms and facilitating the initial steps in drug design.</p>","PeriodicalId":19066,"journal":{"name":"Nature Communications","volume":"129 1","pages":""},"PeriodicalIF":15.7000,"publicationDate":"2025-07-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Sequence-based virtual screening using transformers\",\"authors\":\"Shengyu Zhang, Donghui Huo, Robert I. Horne, Yumeng Qi, Sebastian Pujalte Ojeda, Aixia Yan, Michele Vendruscolo\",\"doi\":\"10.1038/s41467-025-61833-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Protein-ligand interactions play central roles in myriad biological processes and are of key importance in drug design. Deep learning approaches are becoming cost-effective alternatives to high-throughput experimental methods for ligand identification. Here, to predict the binding affinity between proteins and small molecules, we introduce Ligand-Transformer, a deep learning method based on the transformer architecture. Ligand-Transformer implements a sequence-based approach, where the inputs are the amino acid sequence of the target protein and the topology of the small molecule to enable the prediction of the conformational space explored by the complex between the two. We apply Ligand-Transformer to screen and validate experimentally inhibitors targeting the mutant EGFR<sup>LTC</sup> kinase, identifying compounds with low nanomolar potency. We then use this approach to predict the conformational population shifts induced by known ABL kinase inhibitors, showing that sequence-based predictions enable the characterisation of the population shift upon binding. Overall, our results illustrate the potential of Ligand-Transformer to accurately predict the interactions of small molecules with proteins, including the binding affinity and the changes in the free energy landscapes upon binding, thus uncovering molecular mechanisms and facilitating the initial steps in drug design.</p>\",\"PeriodicalId\":19066,\"journal\":{\"name\":\"Nature Communications\",\"volume\":\"129 1\",\"pages\":\"\"},\"PeriodicalIF\":15.7000,\"publicationDate\":\"2025-07-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature Communications\",\"FirstCategoryId\":\"103\",\"ListUrlMain\":\"https://doi.org/10.1038/s41467-025-61833-8\",\"RegionNum\":1,\"RegionCategory\":\"综合性期刊\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MULTIDISCIPLINARY SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Communications","FirstCategoryId":"103","ListUrlMain":"https://doi.org/10.1038/s41467-025-61833-8","RegionNum":1,"RegionCategory":"综合性期刊","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
Sequence-based virtual screening using transformers
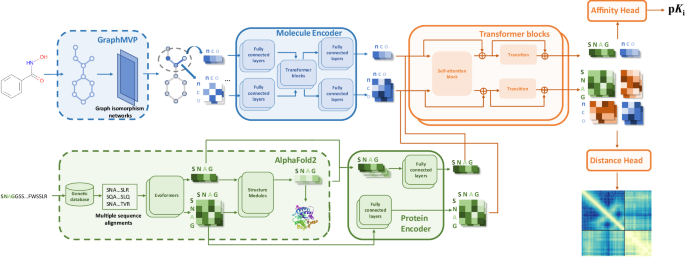
Protein-ligand interactions play central roles in myriad biological processes and are of key importance in drug design. Deep learning approaches are becoming cost-effective alternatives to high-throughput experimental methods for ligand identification. Here, to predict the binding affinity between proteins and small molecules, we introduce Ligand-Transformer, a deep learning method based on the transformer architecture. Ligand-Transformer implements a sequence-based approach, where the inputs are the amino acid sequence of the target protein and the topology of the small molecule to enable the prediction of the conformational space explored by the complex between the two. We apply Ligand-Transformer to screen and validate experimentally inhibitors targeting the mutant EGFRLTC kinase, identifying compounds with low nanomolar potency. We then use this approach to predict the conformational population shifts induced by known ABL kinase inhibitors, showing that sequence-based predictions enable the characterisation of the population shift upon binding. Overall, our results illustrate the potential of Ligand-Transformer to accurately predict the interactions of small molecules with proteins, including the binding affinity and the changes in the free energy landscapes upon binding, thus uncovering molecular mechanisms and facilitating the initial steps in drug design.
期刊介绍:
Nature Communications, an open-access journal, publishes high-quality research spanning all areas of the natural sciences. Papers featured in the journal showcase significant advances relevant to specialists in each respective field. With a 2-year impact factor of 16.6 (2022) and a median time of 8 days from submission to the first editorial decision, Nature Communications is committed to rapid dissemination of research findings. As a multidisciplinary journal, it welcomes contributions from biological, health, physical, chemical, Earth, social, mathematical, applied, and engineering sciences, aiming to highlight important breakthroughs within each domain.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


