Dipankar De, Akash P Mustari, Debajyoti Chatterjee, Rahul Mahajan, Vinod Kumar, Sanjeev Handa
{"title":"类天疱疮扁平苔藓:12例印度患者的临床、组织病理学和免疫学报告。","authors":"Dipankar De, Akash P Mustari, Debajyoti Chatterjee, Rahul Mahajan, Vinod Kumar, Sanjeev Handa","doi":"10.4103/idoj.idoj_763_24","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Lichen planus pemphigoides (LPP) is a rare autoimmune subepidermal blistering disease presenting with lichenoid papules and plaques and tense blisters. There is a paucity of literature on LPP globally.</p><p><strong>Objective: </strong>To report the clinico-demographic profile, histopathology, immunological features, and associated comorbidities in LPP patients.</p><p><strong>Patients and methods: </strong>This was a retrospective study, where past records of all LPP patients diagnosed and treated between November 2013 and October 2022 were included. Patients having a compatible clinical presentation with histopathological and immunological (direct immunofluorescence) evidence of LPP were included.</p><p><strong>Results: </strong>There were 12 LPP patients, with a female-to-male ratio of 2:1. The mean age at diagnosis was 49.6 years and the mean duration of illness before presentation was 3.1 years. Clinical presentation included tense blisters and lichenoid lesions. Oral mucosal involvement was seen in six (50%) patients. Comorbidities were present in three patients. Histopathology showed a subepidermal split in 10 (83.3%), basal cell damage and pigment incontinence in four (33.3%), hypergranulosis and apoptotic keratinocytes in two (16.7%), and lichenoid infiltrate in papillary dermis in one (8.3%) patient. Perilesional direct immunofluorescence (DIF) revealed linear deposits of immunoglobulin G (IgG) and complement component 3 (C3) at the dermo-epidermal junction. The salt-split indirect immunofluorescence done in three patients showed roof binding. Enzyme-linked immunosorbent assay (ELISA) done in three patients showed antibodies against BP180. The majority of patients (83.3%) were treated with oral prednisolone, either alone (16.7%) or in combination (83.3%) with adjuvants.</p><p><strong>Limitations: </strong>Retrospective design and small sample size are the limitations.</p><p><strong>Conclusion: </strong>LPP is a rare subepidermal blistering disorder seen more commonly in adult females. DIF, ELISA, and salt-split indirect immunofluorescence are helpful tools in confirming the diagnosis of LPP and differentiating from bullous lichen planus. Oral corticosteroids comprised the mainstay of therapy. Azathioprine or dapsone were commonly used adjuvants.</p>","PeriodicalId":13335,"journal":{"name":"Indian Dermatology Online Journal","volume":" ","pages":"751-754"},"PeriodicalIF":2.0000,"publicationDate":"2025-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12419719/pdf/","citationCount":"0","resultStr":"{\"title\":\"Lichen Planus Pemphigoides: A Clinical, Histopathological, and Immunological Report of 12 Indian Patients.\",\"authors\":\"Dipankar De, Akash P Mustari, Debajyoti Chatterjee, Rahul Mahajan, Vinod Kumar, Sanjeev Handa\",\"doi\":\"10.4103/idoj.idoj_763_24\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Lichen planus pemphigoides (LPP) is a rare autoimmune subepidermal blistering disease presenting with lichenoid papules and plaques and tense blisters. There is a paucity of literature on LPP globally.</p><p><strong>Objective: </strong>To report the clinico-demographic profile, histopathology, immunological features, and associated comorbidities in LPP patients.</p><p><strong>Patients and methods: </strong>This was a retrospective study, where past records of all LPP patients diagnosed and treated between November 2013 and October 2022 were included. Patients having a compatible clinical presentation with histopathological and immunological (direct immunofluorescence) evidence of LPP were included.</p><p><strong>Results: </strong>There were 12 LPP patients, with a female-to-male ratio of 2:1. The mean age at diagnosis was 49.6 years and the mean duration of illness before presentation was 3.1 years. Clinical presentation included tense blisters and lichenoid lesions. Oral mucosal involvement was seen in six (50%) patients. Comorbidities were present in three patients. Histopathology showed a subepidermal split in 10 (83.3%), basal cell damage and pigment incontinence in four (33.3%), hypergranulosis and apoptotic keratinocytes in two (16.7%), and lichenoid infiltrate in papillary dermis in one (8.3%) patient. Perilesional direct immunofluorescence (DIF) revealed linear deposits of immunoglobulin G (IgG) and complement component 3 (C3) at the dermo-epidermal junction. The salt-split indirect immunofluorescence done in three patients showed roof binding. Enzyme-linked immunosorbent assay (ELISA) done in three patients showed antibodies against BP180. The majority of patients (83.3%) were treated with oral prednisolone, either alone (16.7%) or in combination (83.3%) with adjuvants.</p><p><strong>Limitations: </strong>Retrospective design and small sample size are the limitations.</p><p><strong>Conclusion: </strong>LPP is a rare subepidermal blistering disorder seen more commonly in adult females. DIF, ELISA, and salt-split indirect immunofluorescence are helpful tools in confirming the diagnosis of LPP and differentiating from bullous lichen planus. Oral corticosteroids comprised the mainstay of therapy. Azathioprine or dapsone were commonly used adjuvants.</p>\",\"PeriodicalId\":13335,\"journal\":{\"name\":\"Indian Dermatology Online Journal\",\"volume\":\" \",\"pages\":\"751-754\"},\"PeriodicalIF\":2.0000,\"publicationDate\":\"2025-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12419719/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Indian Dermatology Online Journal\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.4103/idoj.idoj_763_24\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/5/26 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"DERMATOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Indian Dermatology Online Journal","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.4103/idoj.idoj_763_24","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/5/26 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"DERMATOLOGY","Score":null,"Total":0}
Lichen Planus Pemphigoides: A Clinical, Histopathological, and Immunological Report of 12 Indian Patients.
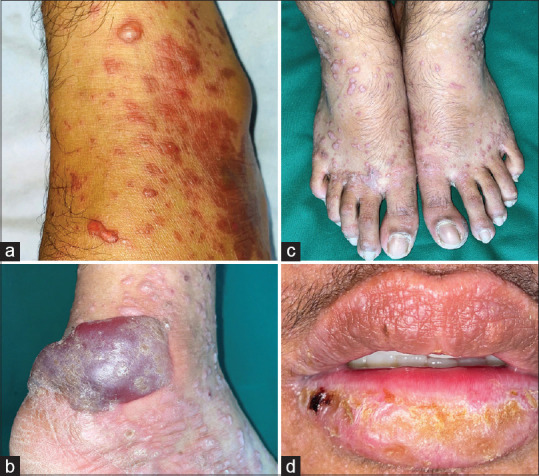
Background: Lichen planus pemphigoides (LPP) is a rare autoimmune subepidermal blistering disease presenting with lichenoid papules and plaques and tense blisters. There is a paucity of literature on LPP globally.
Objective: To report the clinico-demographic profile, histopathology, immunological features, and associated comorbidities in LPP patients.
Patients and methods: This was a retrospective study, where past records of all LPP patients diagnosed and treated between November 2013 and October 2022 were included. Patients having a compatible clinical presentation with histopathological and immunological (direct immunofluorescence) evidence of LPP were included.
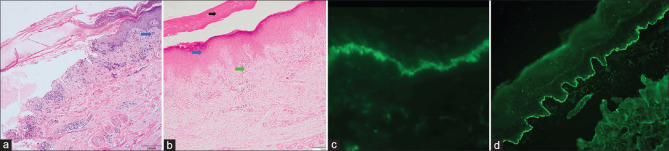
Results: There were 12 LPP patients, with a female-to-male ratio of 2:1. The mean age at diagnosis was 49.6 years and the mean duration of illness before presentation was 3.1 years. Clinical presentation included tense blisters and lichenoid lesions. Oral mucosal involvement was seen in six (50%) patients. Comorbidities were present in three patients. Histopathology showed a subepidermal split in 10 (83.3%), basal cell damage and pigment incontinence in four (33.3%), hypergranulosis and apoptotic keratinocytes in two (16.7%), and lichenoid infiltrate in papillary dermis in one (8.3%) patient. Perilesional direct immunofluorescence (DIF) revealed linear deposits of immunoglobulin G (IgG) and complement component 3 (C3) at the dermo-epidermal junction. The salt-split indirect immunofluorescence done in three patients showed roof binding. Enzyme-linked immunosorbent assay (ELISA) done in three patients showed antibodies against BP180. The majority of patients (83.3%) were treated with oral prednisolone, either alone (16.7%) or in combination (83.3%) with adjuvants.
Limitations: Retrospective design and small sample size are the limitations.
Conclusion: LPP is a rare subepidermal blistering disorder seen more commonly in adult females. DIF, ELISA, and salt-split indirect immunofluorescence are helpful tools in confirming the diagnosis of LPP and differentiating from bullous lichen planus. Oral corticosteroids comprised the mainstay of therapy. Azathioprine or dapsone were commonly used adjuvants.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


