EnsembleFlex:蛋白质结构集成分析变得容易
IF 4.3
2区 生物学
Q2 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
摘要
蛋白质表现出支撑功能的构象动力学,并为药物设计提供信息。EnsembleFlex是一个计算套件,用于从实验确定的结构集合中提取,量化和可视化构象异质性,从而使计算和实验科学家能够获得对蛋白质动力学,配体相互作用和药物设计应用的可操作见解。它通过优化的叠加、降维(主成分分析[PCA]和均匀流形近似和投影[UMAP])、聚类、自动结合位点频率映射和保守水检测来执行双尺度柔性分析(主链和侧链)。可通过无代码图形界面或可脚本化管道访问,EnsembleFlex接受异构PDB集(x射线,核磁共振和低温电子显微镜[cro - em]),并可选择集成弹性网络模型预测(各向异性网络模型[ANM]/高斯网络模型[GNM])来补充实验数据。案例研究——包括腺苷酸激酶、己糖激酶-1、白介素-1β (IL-1β)片段筛选和SARS-CoV-2主要蛋白酶组合——证明了其可扩展性和高通量结构分析的实用性。本文章由计算机程序翻译,如有差异,请以英文原文为准。
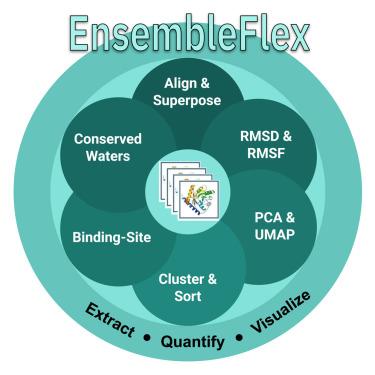
EnsembleFlex: Protein structure ensemble analysis made easy
Proteins exhibit conformational dynamics that underpin function and inform drug design. EnsembleFlex is a computational suite to extract, quantify, and visualize conformational heterogeneity from experimentally determined structure ensembles, thereby enabling both computational and experimental scientists to gain actionable insights into protein dynamics, ligand interactions, and drug-design applications. It performs dual-scale flexibility analysis (backbone and side-chain) via optimized superposition, dimension reduction (principal-component analysis [PCA] and uniform manifold approximation and projection [UMAP]), clustering, automated binding-site frequency mapping, and conserved water detection. Accessible through a no-code graphical interface or scriptable pipelines, EnsembleFlex accepts heterogeneous PDB sets (X-ray, NMR, and cryoelectron microscopy [cryo-EM]) and optionally integrates elastic network-model predictions (anisotropic network model [ANM]/Gaussian network model [GNM]) to complement experimental data. Case studies—including adenylate kinase, hexokinase-1, interleukin-1β (IL-1β) fragment screens, and SARS-CoV-2 main protease ensembles—demonstrate its scalability and utility for high-throughput structural analysis.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Structure
生物-生化与分子生物学
CiteScore
8.90
自引率
1.80%
发文量
155
审稿时长
3-8 weeks
期刊介绍:
Structure aims to publish papers of exceptional interest in the field of structural biology. The journal strives to be essential reading for structural biologists, as well as biologists and biochemists that are interested in macromolecular structure and function. Structure strongly encourages the submission of manuscripts that present structural and molecular insights into biological function and mechanism. Other reports that address fundamental questions in structural biology, such as structure-based examinations of protein evolution, folding, and/or design, will also be considered. We will consider the application of any method, experimental or computational, at high or low resolution, to conduct structural investigations, as long as the method is appropriate for the biological, functional, and mechanistic question(s) being addressed. Likewise, reports describing single-molecule analysis of biological mechanisms are welcome.
In general, the editors encourage submission of experimental structural studies that are enriched by an analysis of structure-activity relationships and will not consider studies that solely report structural information unless the structure or analysis is of exceptional and broad interest. Studies reporting only homology models, de novo models, or molecular dynamics simulations are also discouraged unless the models are informed by or validated by novel experimental data; rationalization of a large body of existing experimental evidence and making testable predictions based on a model or simulation is often not considered sufficient.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


