基于长读测序的1019种不同人类的结构变异。
IF 48.5
1区 综合性期刊
Q1 MULTIDISCIPLINARY SCIENCES
引用次数: 0
摘要
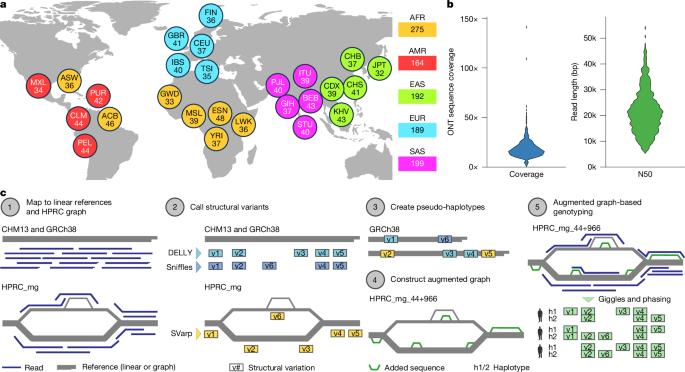
基因组结构变异(SVs)在遗传多样性和人类疾病中起着重要作用1-4,但在人群规模队列中仍未得到充分表征5。在这里,我们对1019人进行了长读序列测序,构建了一个覆盖了1000人基因组计划中26个种群的中等覆盖资源。整合线性和基于图谱的基因组分析,我们发现了超过10万个序列分辨的双等位基因SV,并对30万个串联重复序列的多等位基因变量数进行了基因分型,在基于短读的群体规模调查中推进了SV表征。我们描述了不同种群中的缺失、重复、插入和倒位。长分散核元件-1 (L1)和sin - vntr - alu (SVA)的反转录转位活动介导5‘或3’独特序列的转导8,9,具体取决于源移动元件类别和位点。SV断点分析指出了一系列同源介导的过程有助于SV的形成和反复缺失事件。我们的开放获取资源强调了长读测序在推进SV表征方面的价值,并能够指导患者基因组中的变异优先级。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Structural variation in 1,019 diverse humans based on long-read sequencing
Genomic structural variants (SVs) contribute substantially to genetic diversity and human diseases1–4, yet remain under-characterized in population-scale cohorts5. Here we conducted long-read sequencing6 in 1,019 humans to construct an intermediate-coverage resource covering 26 populations from the 1000 Genomes Project. Integrating linear and graph genome-based analyses, we uncover over 100,000 sequence-resolved biallelic SVs and we genotype 300,000 multiallelic variable number of tandem repeats7, advancing SV characterization over short-read-based population-scale surveys3,4. We characterize deletions, duplications, insertions and inversions in distinct populations. Long interspersed nuclear element-1 (L1) and SINE-VNTR-Alu (SVA) retrotransposition activities mediate the transduction8,9 of unique sequence stretches in 5′ or 3′, depending on source mobile element class and locus. SV breakpoint analyses point to a spectrum of homology-mediated processes contributing to SV formation and recurrent deletion events. Our open-access resource underscores the value of long-read sequencing in advancing SV characterization and enables guiding variant prioritization in patient genomes. Intermediate-coverage long-read sequencing in 1,019 diverse humans from the 1000 Genomes Project, representing 26 populations, enables the generation of comprehensive population-scale structural variant catalogues comprising common and rare alleles.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Nature
综合性期刊-综合性期刊
CiteScore
90.00
自引率
1.20%
发文量
3652
审稿时长
3 months
期刊介绍:
Nature is a prestigious international journal that publishes peer-reviewed research in various scientific and technological fields. The selection of articles is based on criteria such as originality, importance, interdisciplinary relevance, timeliness, accessibility, elegance, and surprising conclusions. In addition to showcasing significant scientific advances, Nature delivers rapid, authoritative, insightful news, and interpretation of current and upcoming trends impacting science, scientists, and the broader public. The journal serves a dual purpose: firstly, to promptly share noteworthy scientific advances and foster discussions among scientists, and secondly, to ensure the swift dissemination of scientific results globally, emphasizing their significance for knowledge, culture, and daily life.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


