Ruidong Xiang, Lingzhao Fang, Shuli Liu, George E Liu, Albert Tenesa, Yahui Gao, Brett A Mason, Amanda J Chamberlain, Michael E Goddard
{"title":"遗传评分组学回归和多性状荟萃分析发现了形成牛复合性状的广泛顺式调控效应。","authors":"Ruidong Xiang, Lingzhao Fang, Shuli Liu, George E Liu, Albert Tenesa, Yahui Gao, Brett A Mason, Amanda J Chamberlain, Michael E Goddard","doi":"10.1093/pnasnexus/pgaf208","DOIUrl":null,"url":null,"abstract":"<p><p>To complete the genome-to-phenome map, transcriptome-wide association studies (TWAS) are performed to correlate genetically predicted gene expression with observed phenotypic measurements. However, the relatively small training population assayed with gene expression could limit the accuracy of TWAS. We propose genetic score omics regression (GSOR) correlating observed gene expression with genetically predicted phenotype, i.e. estimated breeding values (EBVs) in agriculture or polygenic score (PGS) in medicine. The score, calculated using variants near genes with assayed expression (<i>cis</i>-EBV or <i>cis</i>-PGS), provides a powerful association test between <i>cis-</i>effects on gene expression and the trait. In simulated and real data, GSOR outperforms TWAS in detecting causal/informative genes. We applied GSOR to transcriptomes of 16 tissues (<i>N</i> ∼ 5,000) and 37 traits in ∼120,000 cattle and conducted multitrait meta-analyses of omics-associations (MTAO). We found that, on average, each significant gene expression and splicing mediates <i>cis</i>-genetic effects on 8-10 traits. Many prioritized genes by GSOR and MTAO can be verified by Mendelian randomization analysis and show significantly reduced d<i>N</i>/d<i>S</i>, suggesting elevated evolutionary constraint for these genes. Using multiple methods, we detect expression levels of genes and/or RNA splicing events underlying previously thought single-gene loci to influence multiple traits. For example, the expression and RNA splicing of <i>DGAT1</i> from multiple tissues regulated milk production, mastitis, gestation length, temperament, and stature. Also, gene expression and splicing of <i>ABO</i> (Histo-blood group) and <i>ACHE</i> (acetylcholinesterase, Cartwright blood group) affected protein concentration and mastitis, respectively. Taken together, our work provides new methods and biological insights for prioritizing informative omics-phenotype associations in mammals.</p>","PeriodicalId":74468,"journal":{"name":"PNAS nexus","volume":"4 7","pages":"pgaf208"},"PeriodicalIF":3.8000,"publicationDate":"2025-07-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12275098/pdf/","citationCount":"0","resultStr":"{\"title\":\"Genetic score omics regression and multitrait meta-analysis detect widespread <i>cis</i>-regulatory effects shaping bovine complex traits.\",\"authors\":\"Ruidong Xiang, Lingzhao Fang, Shuli Liu, George E Liu, Albert Tenesa, Yahui Gao, Brett A Mason, Amanda J Chamberlain, Michael E Goddard\",\"doi\":\"10.1093/pnasnexus/pgaf208\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>To complete the genome-to-phenome map, transcriptome-wide association studies (TWAS) are performed to correlate genetically predicted gene expression with observed phenotypic measurements. However, the relatively small training population assayed with gene expression could limit the accuracy of TWAS. We propose genetic score omics regression (GSOR) correlating observed gene expression with genetically predicted phenotype, i.e. estimated breeding values (EBVs) in agriculture or polygenic score (PGS) in medicine. The score, calculated using variants near genes with assayed expression (<i>cis</i>-EBV or <i>cis</i>-PGS), provides a powerful association test between <i>cis-</i>effects on gene expression and the trait. In simulated and real data, GSOR outperforms TWAS in detecting causal/informative genes. We applied GSOR to transcriptomes of 16 tissues (<i>N</i> ∼ 5,000) and 37 traits in ∼120,000 cattle and conducted multitrait meta-analyses of omics-associations (MTAO). We found that, on average, each significant gene expression and splicing mediates <i>cis</i>-genetic effects on 8-10 traits. Many prioritized genes by GSOR and MTAO can be verified by Mendelian randomization analysis and show significantly reduced d<i>N</i>/d<i>S</i>, suggesting elevated evolutionary constraint for these genes. Using multiple methods, we detect expression levels of genes and/or RNA splicing events underlying previously thought single-gene loci to influence multiple traits. For example, the expression and RNA splicing of <i>DGAT1</i> from multiple tissues regulated milk production, mastitis, gestation length, temperament, and stature. Also, gene expression and splicing of <i>ABO</i> (Histo-blood group) and <i>ACHE</i> (acetylcholinesterase, Cartwright blood group) affected protein concentration and mastitis, respectively. Taken together, our work provides new methods and biological insights for prioritizing informative omics-phenotype associations in mammals.</p>\",\"PeriodicalId\":74468,\"journal\":{\"name\":\"PNAS nexus\",\"volume\":\"4 7\",\"pages\":\"pgaf208\"},\"PeriodicalIF\":3.8000,\"publicationDate\":\"2025-07-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12275098/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"PNAS nexus\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/pnasnexus/pgaf208\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/7/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"MULTIDISCIPLINARY SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"PNAS nexus","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/pnasnexus/pgaf208","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/7/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
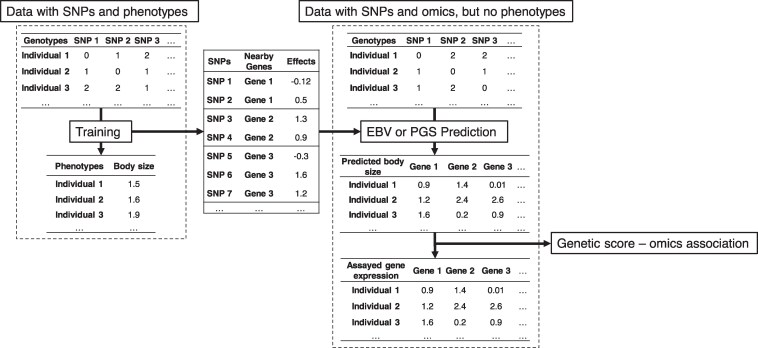
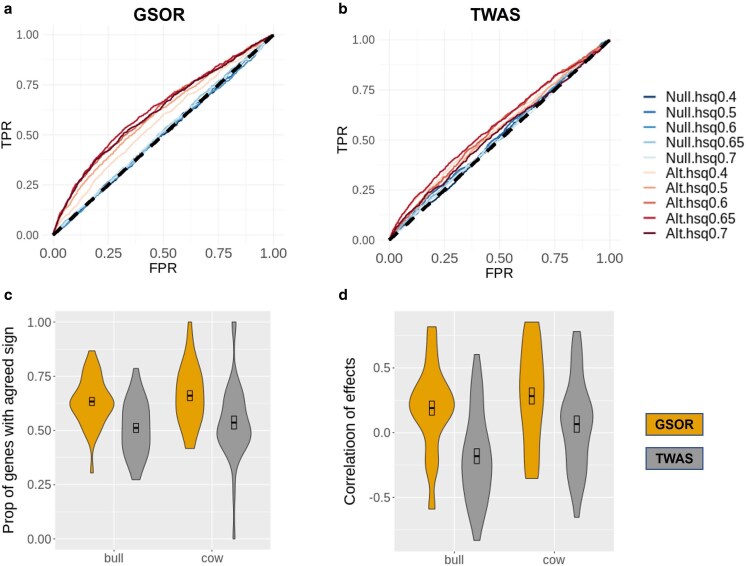
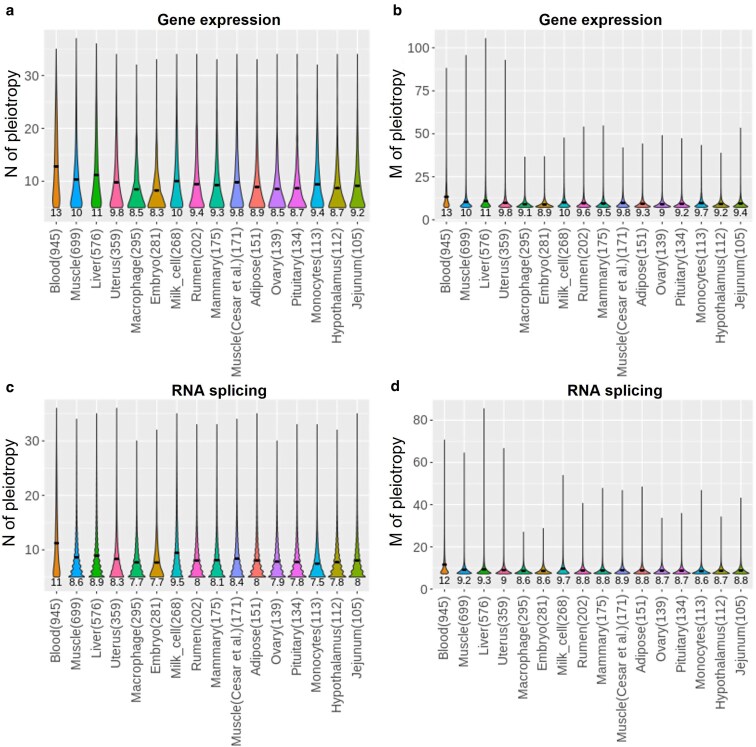
To complete the genome-to-phenome map, transcriptome-wide association studies (TWAS) are performed to correlate genetically predicted gene expression with observed phenotypic measurements. However, the relatively small training population assayed with gene expression could limit the accuracy of TWAS. We propose genetic score omics regression (GSOR) correlating observed gene expression with genetically predicted phenotype, i.e. estimated breeding values (EBVs) in agriculture or polygenic score (PGS) in medicine. The score, calculated using variants near genes with assayed expression (cis-EBV or cis-PGS), provides a powerful association test between cis-effects on gene expression and the trait. In simulated and real data, GSOR outperforms TWAS in detecting causal/informative genes. We applied GSOR to transcriptomes of 16 tissues (N ∼ 5,000) and 37 traits in ∼120,000 cattle and conducted multitrait meta-analyses of omics-associations (MTAO). We found that, on average, each significant gene expression and splicing mediates cis-genetic effects on 8-10 traits. Many prioritized genes by GSOR and MTAO can be verified by Mendelian randomization analysis and show significantly reduced dN/dS, suggesting elevated evolutionary constraint for these genes. Using multiple methods, we detect expression levels of genes and/or RNA splicing events underlying previously thought single-gene loci to influence multiple traits. For example, the expression and RNA splicing of DGAT1 from multiple tissues regulated milk production, mastitis, gestation length, temperament, and stature. Also, gene expression and splicing of ABO (Histo-blood group) and ACHE (acetylcholinesterase, Cartwright blood group) affected protein concentration and mastitis, respectively. Taken together, our work provides new methods and biological insights for prioritizing informative omics-phenotype associations in mammals.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


