{"title":"可解释的机器学习驱动的前列腺癌生物标志物鉴定和验证。","authors":"Jianxu Yuan, Dalin Zhou, Shengjie Yu","doi":"10.21037/tau-2025-242","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Prostate cancer (PCa), a common malignancy among men globally, requires the identification of biomarkers for early diagnosis and predicting progression. This study aimed to identify the key genes involved in the occurrence and development of PCa.</p><p><strong>Methods: </strong>Leveraging data from the Gene Expression Omnibus (GEO) database, this study integrated multi-chip datasets, conducting differential expression analysis and enrichment analysis to pinpoint PCa-related genes. Subsequently, machine learning models were constructed using least absolute shrinkage and selection operator (LASSO) regression, support vector machine (SVM), and random forest (RF) methods. The optimal model was selected for further study and the contribution of related genes was explained using SHapley Additive exPlanations (SHAP) analysis. Furthermore, gene set enrichment analysis (GSEA) and immune cell infiltration analysis were utilized to uncover the underlying molecular mechanisms.</p><p><strong>Results: </strong>In this study, 222 differentially expressed genes (DEGs) were identified and found to be enriched in functions and pathways potentially associated with PCa. Using multiple machine learning models, eight PCa-related core genes (<i>TRPM4</i>, <i>EDN3</i>, <i>EFCAB4A</i>, <i>FAM83B</i>, <i>PENK</i>, <i>NUDT10</i>, <i>KRT14</i>, and <i>CXCL13</i>) were identified. The most accurate RF model was selected for further study with SHAP analysis, which also revealed the contribution of the above genes. GSEA and immune cell infiltration analysis uncovered distinctions between PCa and normal tissues.</p><p><strong>Conclusions: </strong>This study offered potential biomarkers and a theoretical basis for the diagnosis and treatment for PCa.</p>","PeriodicalId":23270,"journal":{"name":"Translational andrology and urology","volume":"14 6","pages":"1528-1541"},"PeriodicalIF":1.7000,"publicationDate":"2025-06-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12271943/pdf/","citationCount":"0","resultStr":"{\"title\":\"Interpretable machine learning driven biomarker identification and validation for prostate cancer.\",\"authors\":\"Jianxu Yuan, Dalin Zhou, Shengjie Yu\",\"doi\":\"10.21037/tau-2025-242\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Prostate cancer (PCa), a common malignancy among men globally, requires the identification of biomarkers for early diagnosis and predicting progression. This study aimed to identify the key genes involved in the occurrence and development of PCa.</p><p><strong>Methods: </strong>Leveraging data from the Gene Expression Omnibus (GEO) database, this study integrated multi-chip datasets, conducting differential expression analysis and enrichment analysis to pinpoint PCa-related genes. Subsequently, machine learning models were constructed using least absolute shrinkage and selection operator (LASSO) regression, support vector machine (SVM), and random forest (RF) methods. The optimal model was selected for further study and the contribution of related genes was explained using SHapley Additive exPlanations (SHAP) analysis. Furthermore, gene set enrichment analysis (GSEA) and immune cell infiltration analysis were utilized to uncover the underlying molecular mechanisms.</p><p><strong>Results: </strong>In this study, 222 differentially expressed genes (DEGs) were identified and found to be enriched in functions and pathways potentially associated with PCa. Using multiple machine learning models, eight PCa-related core genes (<i>TRPM4</i>, <i>EDN3</i>, <i>EFCAB4A</i>, <i>FAM83B</i>, <i>PENK</i>, <i>NUDT10</i>, <i>KRT14</i>, and <i>CXCL13</i>) were identified. The most accurate RF model was selected for further study with SHAP analysis, which also revealed the contribution of the above genes. GSEA and immune cell infiltration analysis uncovered distinctions between PCa and normal tissues.</p><p><strong>Conclusions: </strong>This study offered potential biomarkers and a theoretical basis for the diagnosis and treatment for PCa.</p>\",\"PeriodicalId\":23270,\"journal\":{\"name\":\"Translational andrology and urology\",\"volume\":\"14 6\",\"pages\":\"1528-1541\"},\"PeriodicalIF\":1.7000,\"publicationDate\":\"2025-06-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12271943/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Translational andrology and urology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.21037/tau-2025-242\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/6/26 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"ANDROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Translational andrology and urology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.21037/tau-2025-242","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/6/26 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"ANDROLOGY","Score":null,"Total":0}
Interpretable machine learning driven biomarker identification and validation for prostate cancer.
Background: Prostate cancer (PCa), a common malignancy among men globally, requires the identification of biomarkers for early diagnosis and predicting progression. This study aimed to identify the key genes involved in the occurrence and development of PCa.
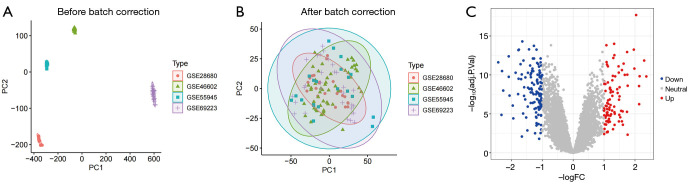
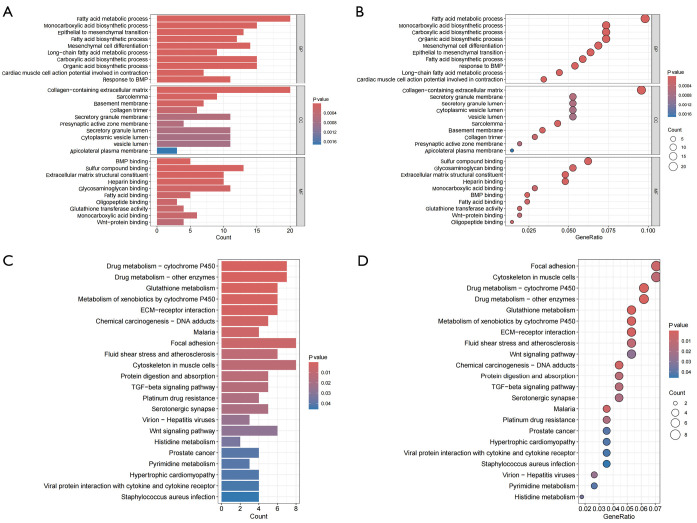
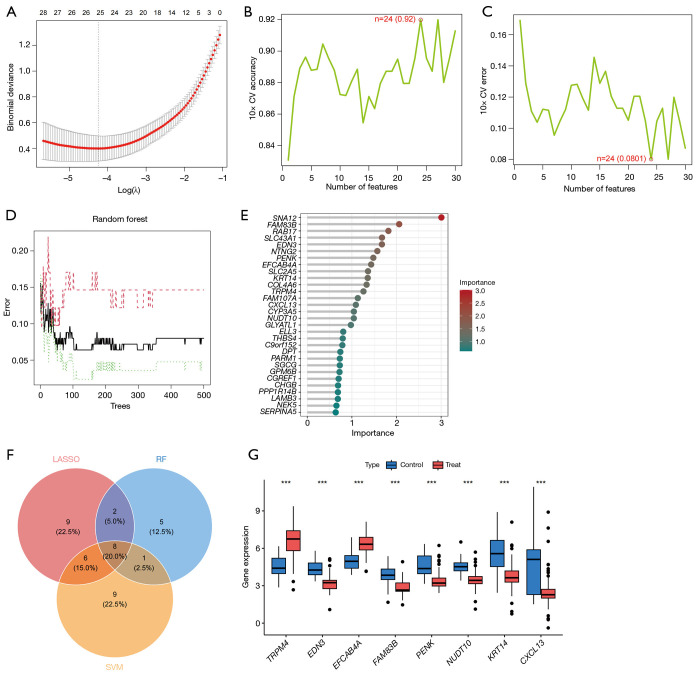
Methods: Leveraging data from the Gene Expression Omnibus (GEO) database, this study integrated multi-chip datasets, conducting differential expression analysis and enrichment analysis to pinpoint PCa-related genes. Subsequently, machine learning models were constructed using least absolute shrinkage and selection operator (LASSO) regression, support vector machine (SVM), and random forest (RF) methods. The optimal model was selected for further study and the contribution of related genes was explained using SHapley Additive exPlanations (SHAP) analysis. Furthermore, gene set enrichment analysis (GSEA) and immune cell infiltration analysis were utilized to uncover the underlying molecular mechanisms.
Results: In this study, 222 differentially expressed genes (DEGs) were identified and found to be enriched in functions and pathways potentially associated with PCa. Using multiple machine learning models, eight PCa-related core genes (TRPM4, EDN3, EFCAB4A, FAM83B, PENK, NUDT10, KRT14, and CXCL13) were identified. The most accurate RF model was selected for further study with SHAP analysis, which also revealed the contribution of the above genes. GSEA and immune cell infiltration analysis uncovered distinctions between PCa and normal tissues.
Conclusions: This study offered potential biomarkers and a theoretical basis for the diagnosis and treatment for PCa.
期刊介绍:
ranslational Andrology and Urology (Print ISSN 2223-4683; Online ISSN 2223-4691; Transl Androl Urol; TAU) is an open access, peer-reviewed, bi-monthly journal (quarterly published from Mar.2012 - Dec. 2014). The main focus of the journal is to describe new findings in the field of translational research of Andrology and Urology, provides current and practical information on basic research and clinical investigations of Andrology and Urology. Specific areas of interest include, but not limited to, molecular study, pathology, biology and technical advances related to andrology and urology. Topics cover range from evaluation, prevention, diagnosis, therapy, prognosis, rehabilitation and future challenges to urology and andrology. Contributions pertinent to urology and andrology are also included from related fields such as public health, basic sciences, education, sociology, and nursing.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


