Zhikai Gao, Zhiguo Wang, Tiren Peng, Xi Sun, Hang Zhang, Zishan Luo, Yuhang Zhou, Lei Zeng, Hong Cui, Weizhi Tian, Rong Feng, Lingxia Jin and Hongkuan Yuan
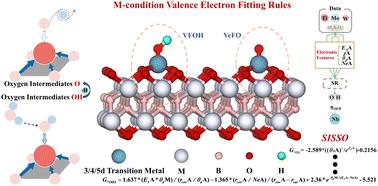
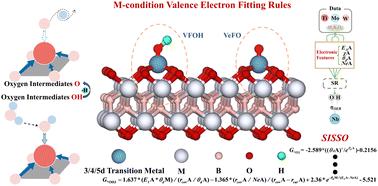
{"title":"基于机器学习的mbe基单原子催化剂界面价电子拟合规律研究","authors":"Zhikai Gao, Zhiguo Wang, Tiren Peng, Xi Sun, Hang Zhang, Zishan Luo, Yuhang Zhou, Lei Zeng, Hong Cui, Weizhi Tian, Rong Feng, Lingxia Jin and Hongkuan Yuan","doi":"10.1039/D5TA04324G","DOIUrl":null,"url":null,"abstract":"<p >Single-atom catalysts (SACs) have garnered significant interest due to their exceptional catalytic activity and selectivity when incorporated into two-dimensional materials. However, the d-band center theory for SACs still exhibits discrepancies in describing the adsorption energies of reaction intermediates. This study integrates machine learning (ML) with density functional theory to introduce a valence electron fitting descriptor for elucidating the adsorption mechanisms of intermediates on MBene-based SACs. By combining DFT calculations with ML-driven feature analysis, an M-condition valence-electron fitting rule (VeFO/VFOH) between the valence electron count of the anchored metal (V<small><sub>TM</sub></small>) and that of the adsorbed intermediates (V<small><sub>O/OH</sub></small>) was identified: M < 5: V<small><sub>O</sub></small> + V<small><sub>TM</sub></small> = 11, V<small><sub>OH</sub></small> + V<small><sub>TM</sub></small> = 11; M = 5: V<small><sub>O</sub></small> + V<small><sub>TM</sub></small> = 12, V<small><sub>OH</sub></small> + V<small><sub>TM</sub></small> = 11; M > 5: V<small><sub>O</sub></small> + V<small><sub>TM</sub></small> = 12, V<small><sub>OH</sub></small> + V<small><sub>TM</sub></small> = 12. This descriptor provides a unified framework for predicting intermediate adsorption behavior across different MBene substrates. Electronic-structure analysis indicates that adsorption is driven by electron-sharing through orbital hybridization, and that optimal orbital resonance positions, pronounced overlap-peak intensities, and moderate charge-transfer magnitudes collectively underpin strong adsorption. Well-fitted multidimensional SISSO adsorption energy descriptors probe the d-electron number of TM and M as the main manifestation of the structure's adsorption capacity, and the structure's ability to adsorb O/OH decreases/increases with increasing d-electron number. The dimensional augmentation of the descriptors enhances the goodness-of-fit (<em>R</em><small><sub>O3</sub></small><small><sup>2</sup></small> = 0.86 and <em>R</em><small><sub>OH3</sub></small><small><sup>2</sup></small> = 0.89) and, concurrently, confirms the validity of the M-conditional valence-electron fitting rule for d-orbital hybridization filling angles. This study reveals the M-conditional valence-electron fitting rule governing adsorption intermediates on TM–M<small><sub>2</sub></small>B<small><sub>2</sub></small>O<small><sub>2</sub></small> materials, thereby rectifying the poor goodness-of-fit exhibited by the conventional d-band center model for adsorption energies (<em>R</em><small><sub>O</sub></small><small><sup>2</sup></small> = 0.02 and <em>R</em><small><sub>OH</sub></small><small><sup>2</sup></small> = 0.25). These insights furnish guidance for the rational design of OER catalysts centered on the OH → O intermediate and establish a novel theoretical framework and design paradigm for understanding and predicting how adsorption energies of reaction intermediates—and their rate-determining conversion steps—vary across different catalytic substrates.</p>","PeriodicalId":82,"journal":{"name":"Journal of Materials Chemistry A","volume":" 32","pages":" 26724-26736"},"PeriodicalIF":9.5000,"publicationDate":"2025-07-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Machine learning-assisted exploration of the interfacial valence electron fitting rule for MBene-based single-atom catalysts†\",\"authors\":\"Zhikai Gao, Zhiguo Wang, Tiren Peng, Xi Sun, Hang Zhang, Zishan Luo, Yuhang Zhou, Lei Zeng, Hong Cui, Weizhi Tian, Rong Feng, Lingxia Jin and Hongkuan Yuan\",\"doi\":\"10.1039/D5TA04324G\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Single-atom catalysts (SACs) have garnered significant interest due to their exceptional catalytic activity and selectivity when incorporated into two-dimensional materials. However, the d-band center theory for SACs still exhibits discrepancies in describing the adsorption energies of reaction intermediates. This study integrates machine learning (ML) with density functional theory to introduce a valence electron fitting descriptor for elucidating the adsorption mechanisms of intermediates on MBene-based SACs. By combining DFT calculations with ML-driven feature analysis, an M-condition valence-electron fitting rule (VeFO/VFOH) between the valence electron count of the anchored metal (V<small><sub>TM</sub></small>) and that of the adsorbed intermediates (V<small><sub>O/OH</sub></small>) was identified: M < 5: V<small><sub>O</sub></small> + V<small><sub>TM</sub></small> = 11, V<small><sub>OH</sub></small> + V<small><sub>TM</sub></small> = 11; M = 5: V<small><sub>O</sub></small> + V<small><sub>TM</sub></small> = 12, V<small><sub>OH</sub></small> + V<small><sub>TM</sub></small> = 11; M > 5: V<small><sub>O</sub></small> + V<small><sub>TM</sub></small> = 12, V<small><sub>OH</sub></small> + V<small><sub>TM</sub></small> = 12. This descriptor provides a unified framework for predicting intermediate adsorption behavior across different MBene substrates. Electronic-structure analysis indicates that adsorption is driven by electron-sharing through orbital hybridization, and that optimal orbital resonance positions, pronounced overlap-peak intensities, and moderate charge-transfer magnitudes collectively underpin strong adsorption. Well-fitted multidimensional SISSO adsorption energy descriptors probe the d-electron number of TM and M as the main manifestation of the structure's adsorption capacity, and the structure's ability to adsorb O/OH decreases/increases with increasing d-electron number. The dimensional augmentation of the descriptors enhances the goodness-of-fit (<em>R</em><small><sub>O3</sub></small><small><sup>2</sup></small> = 0.86 and <em>R</em><small><sub>OH3</sub></small><small><sup>2</sup></small> = 0.89) and, concurrently, confirms the validity of the M-conditional valence-electron fitting rule for d-orbital hybridization filling angles. This study reveals the M-conditional valence-electron fitting rule governing adsorption intermediates on TM–M<small><sub>2</sub></small>B<small><sub>2</sub></small>O<small><sub>2</sub></small> materials, thereby rectifying the poor goodness-of-fit exhibited by the conventional d-band center model for adsorption energies (<em>R</em><small><sub>O</sub></small><small><sup>2</sup></small> = 0.02 and <em>R</em><small><sub>OH</sub></small><small><sup>2</sup></small> = 0.25). These insights furnish guidance for the rational design of OER catalysts centered on the OH → O intermediate and establish a novel theoretical framework and design paradigm for understanding and predicting how adsorption energies of reaction intermediates—and their rate-determining conversion steps—vary across different catalytic substrates.</p>\",\"PeriodicalId\":82,\"journal\":{\"name\":\"Journal of Materials Chemistry A\",\"volume\":\" 32\",\"pages\":\" 26724-26736\"},\"PeriodicalIF\":9.5000,\"publicationDate\":\"2025-07-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Materials Chemistry A\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/ta/d5ta04324g\",\"RegionNum\":2,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Materials Chemistry A","FirstCategoryId":"88","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/ta/d5ta04324g","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Machine learning-assisted exploration of the interfacial valence electron fitting rule for MBene-based single-atom catalysts†
Single-atom catalysts (SACs) have garnered significant interest due to their exceptional catalytic activity and selectivity when incorporated into two-dimensional materials. However, the d-band center theory for SACs still exhibits discrepancies in describing the adsorption energies of reaction intermediates. This study integrates machine learning (ML) with density functional theory to introduce a valence electron fitting descriptor for elucidating the adsorption mechanisms of intermediates on MBene-based SACs. By combining DFT calculations with ML-driven feature analysis, an M-condition valence-electron fitting rule (VeFO/VFOH) between the valence electron count of the anchored metal (VTM) and that of the adsorbed intermediates (VO/OH) was identified: M < 5: VO + VTM = 11, VOH + VTM = 11; M = 5: VO + VTM = 12, VOH + VTM = 11; M > 5: VO + VTM = 12, VOH + VTM = 12. This descriptor provides a unified framework for predicting intermediate adsorption behavior across different MBene substrates. Electronic-structure analysis indicates that adsorption is driven by electron-sharing through orbital hybridization, and that optimal orbital resonance positions, pronounced overlap-peak intensities, and moderate charge-transfer magnitudes collectively underpin strong adsorption. Well-fitted multidimensional SISSO adsorption energy descriptors probe the d-electron number of TM and M as the main manifestation of the structure's adsorption capacity, and the structure's ability to adsorb O/OH decreases/increases with increasing d-electron number. The dimensional augmentation of the descriptors enhances the goodness-of-fit (RO32 = 0.86 and ROH32 = 0.89) and, concurrently, confirms the validity of the M-conditional valence-electron fitting rule for d-orbital hybridization filling angles. This study reveals the M-conditional valence-electron fitting rule governing adsorption intermediates on TM–M2B2O2 materials, thereby rectifying the poor goodness-of-fit exhibited by the conventional d-band center model for adsorption energies (RO2 = 0.02 and ROH2 = 0.25). These insights furnish guidance for the rational design of OER catalysts centered on the OH → O intermediate and establish a novel theoretical framework and design paradigm for understanding and predicting how adsorption energies of reaction intermediates—and their rate-determining conversion steps—vary across different catalytic substrates.
期刊介绍:
The Journal of Materials Chemistry A, B & C covers a wide range of high-quality studies in the field of materials chemistry, with each section focusing on specific applications of the materials studied. Journal of Materials Chemistry A emphasizes applications in energy and sustainability, including topics such as artificial photosynthesis, batteries, and fuel cells. Journal of Materials Chemistry B focuses on applications in biology and medicine, while Journal of Materials Chemistry C covers applications in optical, magnetic, and electronic devices. Example topic areas within the scope of Journal of Materials Chemistry A include catalysis, green/sustainable materials, sensors, and water treatment, among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


