Reem Abdwani, Mahadev J Mal, Eman Al Masroori, Ruqaiya Al Jashmi, Safiya Al Abrawi, Ibrahim Al-Zakwani
{"title":"阿曼青少年皮肌炎:来自国家队列的临床模式和疾病轨迹。","authors":"Reem Abdwani, Mahadev J Mal, Eman Al Masroori, Ruqaiya Al Jashmi, Safiya Al Abrawi, Ibrahim Al-Zakwani","doi":"10.1186/s12969-025-01132-0","DOIUrl":null,"url":null,"abstract":"<p><strong>Objective: </strong>Juvenile dermatomyositis (JDM) is an uncommon autoimmune condition in children, often leading to prolonged disease burden and significant morbidity. Despite global advancements in understanding JDM, studies from the Middle East, particularly Oman, remain scarce. This study aims to characterize JDM from an Omani national cohort, evaluating clinical manifestations, laboratory features, disease course, and treatment outcomes.</p><p><strong>Methods: </strong>A retrospective review of all JDM patients diagnosed and managed by pediatric rheumatologist in tertiary centers in Oman was conducted. Patient demographics, clinical features, laboratory findings, treatment modalities, and disease outcomes were analyzed.</p><p><strong>Results: </strong>A total of 30 children diagnosed with JDM were included. They had an equal female to male distribution, 1:1 ratio. The median age at disease onset was 6.78 years (range: 2-13), with a median diagnostic delay of 8.4 months (range:1-23). The median follow-up period for these patients was 4 years (absolute range: 1 month-16 years). Classic JDM skin manifestations, including heliotrope rash (n = 25; 83%) and Gottron's papules (n = 23; 77%), were common. Proximal muscle weakness was observed in 28 (93%) patients, while 23 (77%) patients exhibited elevated muscle enzymes. MRI findings consistent with myositis were present in 70% (n = 19/27) of the subjects, and muscle biopsy confirmed JDM in 9 cases (30%). Among 25 patients tested for myositis specific antibodies, NXP2 (n = 3), Anti-TIF1 (n = 2), Anti-Mi-2 (n = 1), and MDA5 (n = 1) were detected, showing expected correlations with disease phenotype. Corticosteroids were universally administered, with methotrexate (n = 25; 83%) and IVIG (n = 15; 50%) as common adjuncts. Calcinosis was observed in 8 patients (27%), and was managed with various treatment modalities including pamidronate (n = 3), diltiazem (n = 2), and infliximab (n = 1). At the last follow-up, 18 patients (60%) were in clinical remission, 50% (n = 15) followed a polyphasic or chronic disease course, and 2 patients succumbed to disease-related complications.</p><p><strong>Conclusions: </strong>This study provides comprehensive characterization of pediatric JDM in Oman. The findings highlight regional variations in disease presentation, autoantibody profiles, and treatment responses, underscoring the need for early diagnosis and individualized management strategies. Continued follow-up is essential to optimize long-term outcomes and improve survival rates in this patient population.</p>","PeriodicalId":54630,"journal":{"name":"Pediatric Rheumatology","volume":"23 1","pages":"75"},"PeriodicalIF":2.3000,"publicationDate":"2025-07-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12273291/pdf/","citationCount":"0","resultStr":"{\"title\":\"Juvenile dermatomyositis in Oman: clinical patterns and disease trajectory from a National cohort.\",\"authors\":\"Reem Abdwani, Mahadev J Mal, Eman Al Masroori, Ruqaiya Al Jashmi, Safiya Al Abrawi, Ibrahim Al-Zakwani\",\"doi\":\"10.1186/s12969-025-01132-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Objective: </strong>Juvenile dermatomyositis (JDM) is an uncommon autoimmune condition in children, often leading to prolonged disease burden and significant morbidity. Despite global advancements in understanding JDM, studies from the Middle East, particularly Oman, remain scarce. This study aims to characterize JDM from an Omani national cohort, evaluating clinical manifestations, laboratory features, disease course, and treatment outcomes.</p><p><strong>Methods: </strong>A retrospective review of all JDM patients diagnosed and managed by pediatric rheumatologist in tertiary centers in Oman was conducted. Patient demographics, clinical features, laboratory findings, treatment modalities, and disease outcomes were analyzed.</p><p><strong>Results: </strong>A total of 30 children diagnosed with JDM were included. They had an equal female to male distribution, 1:1 ratio. The median age at disease onset was 6.78 years (range: 2-13), with a median diagnostic delay of 8.4 months (range:1-23). The median follow-up period for these patients was 4 years (absolute range: 1 month-16 years). Classic JDM skin manifestations, including heliotrope rash (n = 25; 83%) and Gottron's papules (n = 23; 77%), were common. Proximal muscle weakness was observed in 28 (93%) patients, while 23 (77%) patients exhibited elevated muscle enzymes. MRI findings consistent with myositis were present in 70% (n = 19/27) of the subjects, and muscle biopsy confirmed JDM in 9 cases (30%). Among 25 patients tested for myositis specific antibodies, NXP2 (n = 3), Anti-TIF1 (n = 2), Anti-Mi-2 (n = 1), and MDA5 (n = 1) were detected, showing expected correlations with disease phenotype. Corticosteroids were universally administered, with methotrexate (n = 25; 83%) and IVIG (n = 15; 50%) as common adjuncts. Calcinosis was observed in 8 patients (27%), and was managed with various treatment modalities including pamidronate (n = 3), diltiazem (n = 2), and infliximab (n = 1). At the last follow-up, 18 patients (60%) were in clinical remission, 50% (n = 15) followed a polyphasic or chronic disease course, and 2 patients succumbed to disease-related complications.</p><p><strong>Conclusions: </strong>This study provides comprehensive characterization of pediatric JDM in Oman. The findings highlight regional variations in disease presentation, autoantibody profiles, and treatment responses, underscoring the need for early diagnosis and individualized management strategies. Continued follow-up is essential to optimize long-term outcomes and improve survival rates in this patient population.</p>\",\"PeriodicalId\":54630,\"journal\":{\"name\":\"Pediatric Rheumatology\",\"volume\":\"23 1\",\"pages\":\"75\"},\"PeriodicalIF\":2.3000,\"publicationDate\":\"2025-07-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12273291/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Pediatric Rheumatology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s12969-025-01132-0\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"PEDIATRICS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Pediatric Rheumatology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12969-025-01132-0","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"PEDIATRICS","Score":null,"Total":0}
Juvenile dermatomyositis in Oman: clinical patterns and disease trajectory from a National cohort.
Objective: Juvenile dermatomyositis (JDM) is an uncommon autoimmune condition in children, often leading to prolonged disease burden and significant morbidity. Despite global advancements in understanding JDM, studies from the Middle East, particularly Oman, remain scarce. This study aims to characterize JDM from an Omani national cohort, evaluating clinical manifestations, laboratory features, disease course, and treatment outcomes.
Methods: A retrospective review of all JDM patients diagnosed and managed by pediatric rheumatologist in tertiary centers in Oman was conducted. Patient demographics, clinical features, laboratory findings, treatment modalities, and disease outcomes were analyzed.
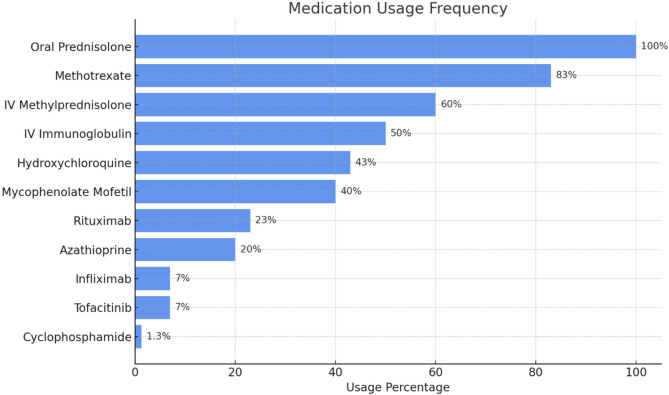
Results: A total of 30 children diagnosed with JDM were included. They had an equal female to male distribution, 1:1 ratio. The median age at disease onset was 6.78 years (range: 2-13), with a median diagnostic delay of 8.4 months (range:1-23). The median follow-up period for these patients was 4 years (absolute range: 1 month-16 years). Classic JDM skin manifestations, including heliotrope rash (n = 25; 83%) and Gottron's papules (n = 23; 77%), were common. Proximal muscle weakness was observed in 28 (93%) patients, while 23 (77%) patients exhibited elevated muscle enzymes. MRI findings consistent with myositis were present in 70% (n = 19/27) of the subjects, and muscle biopsy confirmed JDM in 9 cases (30%). Among 25 patients tested for myositis specific antibodies, NXP2 (n = 3), Anti-TIF1 (n = 2), Anti-Mi-2 (n = 1), and MDA5 (n = 1) were detected, showing expected correlations with disease phenotype. Corticosteroids were universally administered, with methotrexate (n = 25; 83%) and IVIG (n = 15; 50%) as common adjuncts. Calcinosis was observed in 8 patients (27%), and was managed with various treatment modalities including pamidronate (n = 3), diltiazem (n = 2), and infliximab (n = 1). At the last follow-up, 18 patients (60%) were in clinical remission, 50% (n = 15) followed a polyphasic or chronic disease course, and 2 patients succumbed to disease-related complications.
Conclusions: This study provides comprehensive characterization of pediatric JDM in Oman. The findings highlight regional variations in disease presentation, autoantibody profiles, and treatment responses, underscoring the need for early diagnosis and individualized management strategies. Continued follow-up is essential to optimize long-term outcomes and improve survival rates in this patient population.
期刊介绍:
Pediatric Rheumatology is an open access, peer-reviewed, online journal encompassing all aspects of clinical and basic research related to pediatric rheumatology and allied subjects.
The journal’s scope of diseases and syndromes include musculoskeletal pain syndromes, rheumatic fever and post-streptococcal syndromes, juvenile idiopathic arthritis, systemic lupus erythematosus, juvenile dermatomyositis, local and systemic scleroderma, Kawasaki disease, Henoch-Schonlein purpura and other vasculitides, sarcoidosis, inherited musculoskeletal syndromes, autoinflammatory syndromes, and others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


