从蛋白质语言模型到连续结构异质性
IF 4.3
2区 生物学
Q2 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
摘要
蛋白质如何移动和变形决定了它们与环境的相互作用,因此对细胞功能至关重要。随着单蛋白3D结构预测的革命,研究人员专注于重新利用或开发深度学习模型来采样替代蛋白质构象。在这项工作中,我们探索了是否可以直接从序列中预测蛋白质运动的连续紧凑表示,而无需利用3D结构。SeaMoon利用蛋白质语言模型(pLM)嵌入作为轻量级卷积神经网络的输入。我们将SeaMoon与表现出不同运动的~ ~ 1000个实验构象集合进行了评估。对于40%的测试蛋白质,它至少能以合理的精度预测出一次地面真实运动。SeaMoon捕获正常模式分析无法获得的运动,这是一种基于无监督物理的方法,仅依赖于3D几何形状,并将其推广到与训练集没有可检测序列相似性的蛋白质。SeaMoon很容易通过新颖或更新的plm进行再培训。本文章由计算机程序翻译,如有差异,请以英文原文为准。
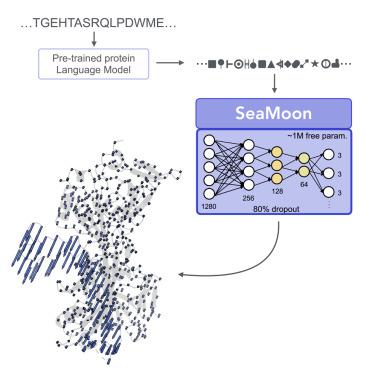
SeaMoon: From protein language models to continuous structural heterogeneity
How proteins move and deform determines their interactions with the environment and is thus of the utmost importance for cellular functioning. Following the revolution in single protein 3D structure prediction, researchers have focused on repurposing or developing deep learning models for sampling alternative protein conformations. In this work, we explored whether continuous compact representations of protein motions could be predicted directly from sequences, without exploiting 3D structures. SeaMoon leverages protein language model (pLM) embeddings as input to a lightweight convolutional neural network. We assessed SeaMoon against 1,000 collections of experimental conformations exhibiting diverse motions. It predicts at least one ground-truth motion with reasonable accuracy for 40% of the test proteins. SeaMoon captures motions inaccessible to normal mode analysis, an unsupervised physics-based method relying solely on 3D geometry, and generalizes to proteins without detectable sequence similarity to the training set. SeaMoon is easily retrainable with novel or updated pLMs.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Structure
生物-生化与分子生物学
CiteScore
8.90
自引率
1.80%
发文量
155
审稿时长
3-8 weeks
期刊介绍:
Structure aims to publish papers of exceptional interest in the field of structural biology. The journal strives to be essential reading for structural biologists, as well as biologists and biochemists that are interested in macromolecular structure and function. Structure strongly encourages the submission of manuscripts that present structural and molecular insights into biological function and mechanism. Other reports that address fundamental questions in structural biology, such as structure-based examinations of protein evolution, folding, and/or design, will also be considered. We will consider the application of any method, experimental or computational, at high or low resolution, to conduct structural investigations, as long as the method is appropriate for the biological, functional, and mechanistic question(s) being addressed. Likewise, reports describing single-molecule analysis of biological mechanisms are welcome.
In general, the editors encourage submission of experimental structural studies that are enriched by an analysis of structure-activity relationships and will not consider studies that solely report structural information unless the structure or analysis is of exceptional and broad interest. Studies reporting only homology models, de novo models, or molecular dynamics simulations are also discouraged unless the models are informed by or validated by novel experimental data; rationalization of a large body of existing experimental evidence and making testable predictions based on a model or simulation is often not considered sufficient.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


