{"title":"UBQLN2在神经退行性疾病中的作用机制和新出现的治疗潜力","authors":"Autumn M Matthews, Alexandra M Whiteley","doi":"10.1042/BST20253053","DOIUrl":null,"url":null,"abstract":"<p><p>Ubiquilins (UBQLNs) regulate cellular protein turnover by shuttling proteins, or 'clients', to the proteasome or autophagy pathways for degradation. Of the five different UBQLN genes in humans, UBQLN2 is the most highly expressed in the nervous system and muscle tissue and has been linked to multiple neurodegenerative diseases. In particular, point mutations of UBQLN2 cause an X-linked, dominant form of amyotrophic lateral sclerosis (ALS), ALS with frontotemporal dementia (ALS/FTD), or FTD. Failed protein degradation is a hallmark of many neurodegenerative diseases, including ALS and FTD; however, it is not clear exactly how ALS/FTD-associated UBQLN2 mutations contribute to pathogenesis. Recent studies have revealed the complexity of UBQLN2 biology and allow deeper understanding as to how UBQLN2 dysfunction may contribute to neurodegenerative disease. UBQLN2 is necessary for mitochondrial protein degradation and for regulating mitochondrial turnover, both of which are essential for motor neurons and have been implicated in the pathogenesis of ALS. Stress granule (SG) formation and regulation are also affected by UBQLN2 mutations, and their dysregulation may contribute to the toxic protein aggregation and SG changes observed in neurodegenerative disease. Finally, there are compelling links connecting UBQLN2 dysfunction with changes to downstream neuronal morphology, function, and behavior. This review will detail the emerging consensus on how UBQLN2 protects against neurodegenerative disease and will provide insights into potential therapeutic approaches.</p>","PeriodicalId":8841,"journal":{"name":"Biochemical Society transactions","volume":" ","pages":"823-833"},"PeriodicalIF":4.3000,"publicationDate":"2025-08-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12410002/pdf/","citationCount":"0","resultStr":"{\"title\":\"UBQLN2 in neurodegenerative disease: mechanistic insights and emerging therapeutic potential.\",\"authors\":\"Autumn M Matthews, Alexandra M Whiteley\",\"doi\":\"10.1042/BST20253053\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Ubiquilins (UBQLNs) regulate cellular protein turnover by shuttling proteins, or 'clients', to the proteasome or autophagy pathways for degradation. Of the five different UBQLN genes in humans, UBQLN2 is the most highly expressed in the nervous system and muscle tissue and has been linked to multiple neurodegenerative diseases. In particular, point mutations of UBQLN2 cause an X-linked, dominant form of amyotrophic lateral sclerosis (ALS), ALS with frontotemporal dementia (ALS/FTD), or FTD. Failed protein degradation is a hallmark of many neurodegenerative diseases, including ALS and FTD; however, it is not clear exactly how ALS/FTD-associated UBQLN2 mutations contribute to pathogenesis. Recent studies have revealed the complexity of UBQLN2 biology and allow deeper understanding as to how UBQLN2 dysfunction may contribute to neurodegenerative disease. UBQLN2 is necessary for mitochondrial protein degradation and for regulating mitochondrial turnover, both of which are essential for motor neurons and have been implicated in the pathogenesis of ALS. Stress granule (SG) formation and regulation are also affected by UBQLN2 mutations, and their dysregulation may contribute to the toxic protein aggregation and SG changes observed in neurodegenerative disease. Finally, there are compelling links connecting UBQLN2 dysfunction with changes to downstream neuronal morphology, function, and behavior. This review will detail the emerging consensus on how UBQLN2 protects against neurodegenerative disease and will provide insights into potential therapeutic approaches.</p>\",\"PeriodicalId\":8841,\"journal\":{\"name\":\"Biochemical Society transactions\",\"volume\":\" \",\"pages\":\"823-833\"},\"PeriodicalIF\":4.3000,\"publicationDate\":\"2025-08-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12410002/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biochemical Society transactions\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1042/BST20253053\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biochemical Society transactions","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1042/BST20253053","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
UBQLN2 in neurodegenerative disease: mechanistic insights and emerging therapeutic potential.
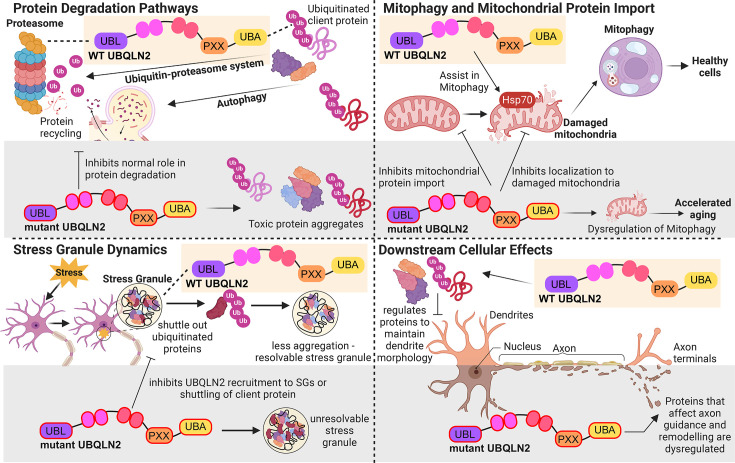
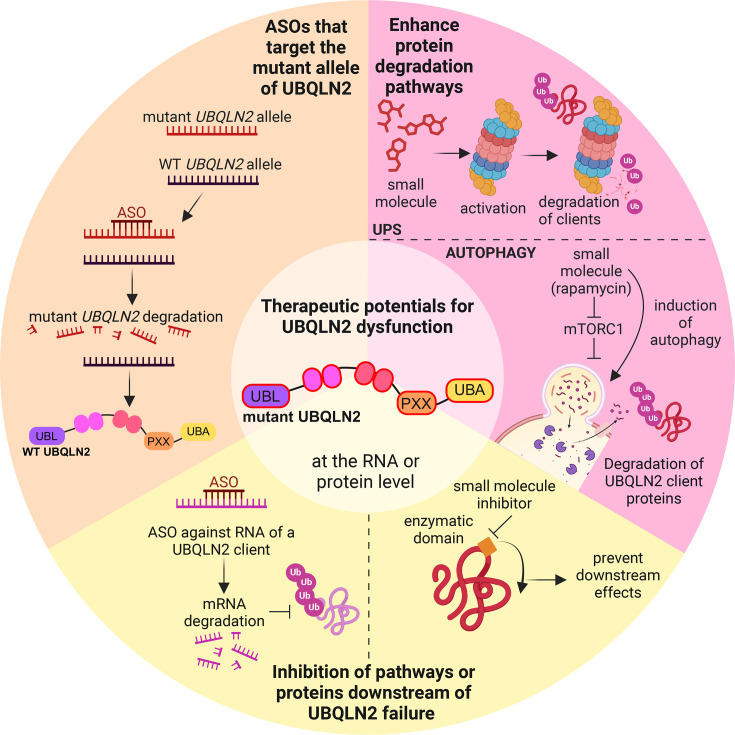
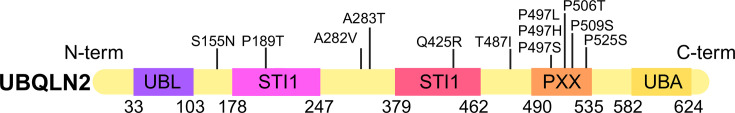
Ubiquilins (UBQLNs) regulate cellular protein turnover by shuttling proteins, or 'clients', to the proteasome or autophagy pathways for degradation. Of the five different UBQLN genes in humans, UBQLN2 is the most highly expressed in the nervous system and muscle tissue and has been linked to multiple neurodegenerative diseases. In particular, point mutations of UBQLN2 cause an X-linked, dominant form of amyotrophic lateral sclerosis (ALS), ALS with frontotemporal dementia (ALS/FTD), or FTD. Failed protein degradation is a hallmark of many neurodegenerative diseases, including ALS and FTD; however, it is not clear exactly how ALS/FTD-associated UBQLN2 mutations contribute to pathogenesis. Recent studies have revealed the complexity of UBQLN2 biology and allow deeper understanding as to how UBQLN2 dysfunction may contribute to neurodegenerative disease. UBQLN2 is necessary for mitochondrial protein degradation and for regulating mitochondrial turnover, both of which are essential for motor neurons and have been implicated in the pathogenesis of ALS. Stress granule (SG) formation and regulation are also affected by UBQLN2 mutations, and their dysregulation may contribute to the toxic protein aggregation and SG changes observed in neurodegenerative disease. Finally, there are compelling links connecting UBQLN2 dysfunction with changes to downstream neuronal morphology, function, and behavior. This review will detail the emerging consensus on how UBQLN2 protects against neurodegenerative disease and will provide insights into potential therapeutic approaches.
期刊介绍:
Biochemical Society Transactions is the reviews journal of the Biochemical Society. Publishing concise reviews written by experts in the field, providing a timely snapshot of the latest developments across all areas of the molecular and cellular biosciences.
Elevating our authors’ ideas and expertise, each review includes a perspectives section where authors offer comment on the latest advances, a glimpse of future challenges and highlighting the importance of associated research areas in far broader contexts.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


