解耦:酸性水氧化过程中钴活性位点的催化降解机制
IF 60.1
1区 材料科学
Q1 ENERGY & FUELS
引用次数: 0
摘要
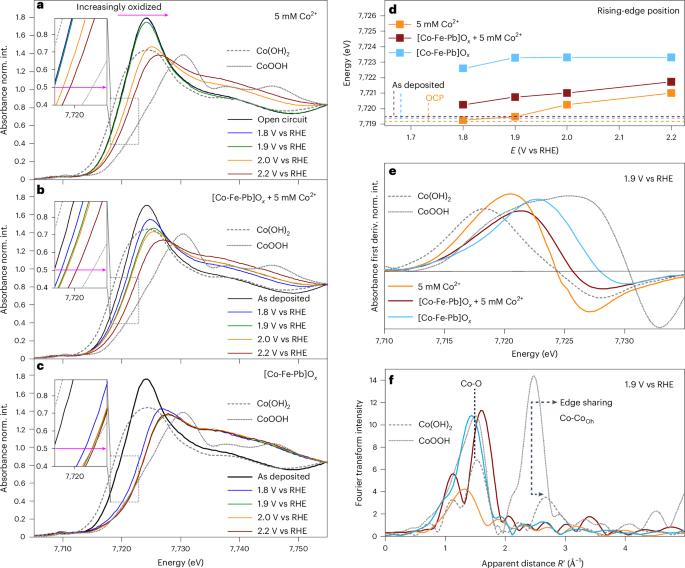
低ph析氧反应(OER)的无铱催化剂的发展是实现千兆瓦级质子交换水电解的必要条件。钴基材料可能满足这一要求,但对这些OER催化剂在低ph下的运行机制知之甚少。在这里,我们研究了活性钴位点的性质和演变,以及电荷和传质过程,这些过程支持它们在钴铁铅氧化物材料中的催化功能,使用原位光谱、重量和电化学技术。我们证明了钴位点的腐蚀及其通过溶解的Co2+的电氧化重组不影响催化机制,并且与OER解耦。超电偶电荷转移由Co(3+δ)+-氧支持,其结构与碱性/近中性条件下的不同,形成时间相对较慢(分钟)。这些机理上的见解可能有助于为这项重要技术开发真正实用的催化剂。本文章由计算机程序翻译,如有差异,请以英文原文为准。

Decoupling the catalytic and degradation mechanisms of cobalt active sites during acidic water oxidation
Advancement of iridium-free catalysts for the low-pH oxygen evolution reaction (OER) is required to enable multi-gigawatt-scale proton-exchange water electrolysis. Cobalt-based materials might address this requirement, but little is known about the mechanism of operation of these OER catalysts at low pH. Here we investigate the nature and evolution of the active cobalt sites along with charge- and mass-transfer processes that support their catalytic function within a cobalt–iron–lead oxide material using in situ spectroscopic, gravimetric and electrochemical techniques. We demonstrate that corrosion of the cobalt sites and their reformation through electrooxidation of dissolved Co2+ do not affect the catalytic mechanism and are decoupled from the OER. The OER-coupled charge transfer is supported by Co(3+δ)+-oxo-species, which are structurally different from those reported for alkaline/near-neutral conditions and are formed on a relatively slow timescale of minutes. These mechanistic insights might assist in developing genuinely practical catalysts for this vital technology. Cobalt–iron–lead oxide electrocatalysts show promise for the low-pH oxygen evolution reaction—an essential reaction in proton-exchange water electrolysis—but can suffer from corrosion. This study uncovers that the mechanism of cobalt site corrosion is decoupled from the oxygen evolution reaction, paving the way for more stable catalyst designs.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Nature Energy
Energy-Energy Engineering and Power Technology
CiteScore
75.10
自引率
1.10%
发文量
193
期刊介绍:
Nature Energy is a monthly, online-only journal committed to showcasing the most impactful research on energy, covering everything from its generation and distribution to the societal implications of energy technologies and policies.
With a focus on exploring all facets of the ongoing energy discourse, Nature Energy delves into topics such as energy generation, storage, distribution, management, and the societal impacts of energy technologies and policies. Emphasizing studies that push the boundaries of knowledge and contribute to the development of next-generation solutions, the journal serves as a platform for the exchange of ideas among stakeholders at the forefront of the energy sector.
Maintaining the hallmark standards of the Nature brand, Nature Energy boasts a dedicated team of professional editors, a rigorous peer-review process, meticulous copy-editing and production, rapid publication times, and editorial independence.
In addition to original research articles, Nature Energy also publishes a range of content types, including Comments, Perspectives, Reviews, News & Views, Features, and Correspondence, covering a diverse array of disciplines relevant to the field of energy.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


