{"title":"基于分子模拟的深度神经网络模型解析5-氟尿嘧啶在COFs中的吸附","authors":"Khushboo Yadava and Ashutosh Yadav","doi":"10.1039/D5PM00007F","DOIUrl":null,"url":null,"abstract":"<p >A database of 1242 experimentally synthesized COFs has been studied to understand their potential as drug carriers by employing molecular simulations and machine learning models to analyze the adsorption abilities and predict the capacity of loading the anticancer drug, 5-fluorouracil. Our findings indicate that different organic linkers, structural features, binding sites, topologies, <em>etc.</em> of COFs play an important role in determining the maximum loading capacity and release parameters of 5-FU. The implementation of molecular simulations-based machine learning methods for drug adsorption studies in COFs is rare in the literature. Once the model was validated, we studied the maximum loading capacity of 5-FU in a series of COFs, 102–108 and 112, from the COF database, as these exhibited a gradual trend in textural properties, aiming to understand this trend and the correlation between their structure and loading capacity. Then, we proceeded to study the adsorption process in detail in 4 of the COFs: three 2D COFs—COF-206, <em>i.e.</em>, D<small><sub>CuPc</sub></small>–A<small><sub>NDI</sub></small>-COF; COF-362, <em>i.e.</em>, PI-COF-3; and COF-398, <em>i.e.</em>, Py-DBA-COF-1—and one 3D COF—COF-363, <em>i.e.</em>, PI-COF-4. Radial distribution function and adsorption energy analyses revealed some important interactions and thermodynamic parameters leading to strong binding and slow release of 5-FU. The adsorption energy values in the top-performing COFs fall within the range of −8.43 to −42.25 × 10<small><sup>3</sup></small> kJ mol<small><sup>−1</sup></small>. The correlation of ML input parameters in terms of various chemical and structural descriptors with the maximum loading capacity is discussed. From the molecular simulations, COF-362 is the best-performing COF in terms of loading capacity and adsorption energy values. The ML models, <em>i.e.</em>, random forest, decision tree and three deep neural networks, were trained on 80% of the total data, while the remaining 20% of the data was used to test the models. DNN model-3 was chosen as the final model for further analysis based on <em>R</em><small><sup>2</sup></small> = 0.87, RMSE = 189.81, and MAE = 100.87. SHapley Additive exPlanations (SHAP) analysis and the feature importance chart indicated that among the structural descriptors, <em>S</em><small><sub>acc</sub></small>, LCD, and <em>V</em><small><sub>f</sub></small>, and among the chemical descriptors, C, H, and N, had the most positive impact on the output predictions of the model. Finally, a graphical user interface based on the best-performing ML model was created to predict the 5-FU loading capacity of COFs. This will save users time without the need to run the code or perform various tedious drug-loading experiments.</p>","PeriodicalId":101141,"journal":{"name":"RSC Pharmaceutics","volume":" 4","pages":" 703-717"},"PeriodicalIF":0.0000,"publicationDate":"2025-04-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2025/pm/d5pm00007f?page=search","citationCount":"0","resultStr":"{\"title\":\"A molecular simulation-based deep neural network model for deciphering the adsorption of 5-Fluorouracil in COFs†\",\"authors\":\"Khushboo Yadava and Ashutosh Yadav\",\"doi\":\"10.1039/D5PM00007F\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >A database of 1242 experimentally synthesized COFs has been studied to understand their potential as drug carriers by employing molecular simulations and machine learning models to analyze the adsorption abilities and predict the capacity of loading the anticancer drug, 5-fluorouracil. Our findings indicate that different organic linkers, structural features, binding sites, topologies, <em>etc.</em> of COFs play an important role in determining the maximum loading capacity and release parameters of 5-FU. The implementation of molecular simulations-based machine learning methods for drug adsorption studies in COFs is rare in the literature. Once the model was validated, we studied the maximum loading capacity of 5-FU in a series of COFs, 102–108 and 112, from the COF database, as these exhibited a gradual trend in textural properties, aiming to understand this trend and the correlation between their structure and loading capacity. Then, we proceeded to study the adsorption process in detail in 4 of the COFs: three 2D COFs—COF-206, <em>i.e.</em>, D<small><sub>CuPc</sub></small>–A<small><sub>NDI</sub></small>-COF; COF-362, <em>i.e.</em>, PI-COF-3; and COF-398, <em>i.e.</em>, Py-DBA-COF-1—and one 3D COF—COF-363, <em>i.e.</em>, PI-COF-4. Radial distribution function and adsorption energy analyses revealed some important interactions and thermodynamic parameters leading to strong binding and slow release of 5-FU. The adsorption energy values in the top-performing COFs fall within the range of −8.43 to −42.25 × 10<small><sup>3</sup></small> kJ mol<small><sup>−1</sup></small>. The correlation of ML input parameters in terms of various chemical and structural descriptors with the maximum loading capacity is discussed. From the molecular simulations, COF-362 is the best-performing COF in terms of loading capacity and adsorption energy values. The ML models, <em>i.e.</em>, random forest, decision tree and three deep neural networks, were trained on 80% of the total data, while the remaining 20% of the data was used to test the models. DNN model-3 was chosen as the final model for further analysis based on <em>R</em><small><sup>2</sup></small> = 0.87, RMSE = 189.81, and MAE = 100.87. SHapley Additive exPlanations (SHAP) analysis and the feature importance chart indicated that among the structural descriptors, <em>S</em><small><sub>acc</sub></small>, LCD, and <em>V</em><small><sub>f</sub></small>, and among the chemical descriptors, C, H, and N, had the most positive impact on the output predictions of the model. Finally, a graphical user interface based on the best-performing ML model was created to predict the 5-FU loading capacity of COFs. This will save users time without the need to run the code or perform various tedious drug-loading experiments.</p>\",\"PeriodicalId\":101141,\"journal\":{\"name\":\"RSC Pharmaceutics\",\"volume\":\" 4\",\"pages\":\" 703-717\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2025-04-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2025/pm/d5pm00007f?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"RSC Pharmaceutics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/pm/d5pm00007f\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"RSC Pharmaceutics","FirstCategoryId":"1085","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/pm/d5pm00007f","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
摘要
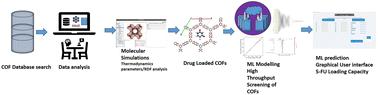
通过分子模拟和机器学习模型分析COFs的吸附能力,并预测其装载抗癌药物5-氟尿嘧啶的能力,研究了1242个实验合成COFs的数据库,以了解它们作为药物载体的潜力。我们的研究结果表明,不同的有机连接体、结构特征、结合位点、拓扑结构等对5-FU的最大负载能力和释放参数起着重要作用。基于分子模拟的机器学习方法在COFs中进行药物吸附研究在文献中是罕见的。模型验证后,我们从COF数据库中研究了一系列COFs(102-108和112)中5-FU的最大承载能力,因为这些COFs在纹理性能上呈现渐变趋势,旨在了解这种趋势以及它们的结构与承载能力之间的相关性。然后,我们对4种COFs的吸附过程进行了详细的研究:3种2D COFs - cof -206,即dcup - and i - cof;COF-362,即PI-COF-3;和COF-398,即py - dba - cof -1和一个3D COF-COF-363,即PI-COF-4。径向分布函数和吸附能分析揭示了导致5-FU强结合和缓释的重要相互作用和热力学参数。性能最好的COFs的吸附能在−8.43 ~−42.25 × 103 kJ mol−1之间。讨论了基于各种化学和结构描述符的机器学习输入参数与最大承载能力的相关性。从分子模拟来看,COF-362在负载能力和吸附能值方面表现最好。ML模型,即随机森林、决策树和三个深度神经网络,在80%的总数据上进行训练,而剩下的20%的数据用于测试模型。根据R2 = 0.87, RMSE = 189.81, MAE = 100.87,最终选择DNN model-3作为进一步分析的最终模型。SHapley加性解释(SHAP)分析和特征重要性图表明,在结构描述符中Sacc、LCD和Vf,在化学描述符中C、H和N对模型的输出预测有最积极的影响。最后,基于最佳ML模型创建了一个图形用户界面来预测COFs的5-FU负载能力。这将节省用户的时间,而无需运行代码或执行各种繁琐的药物加载实验。
A molecular simulation-based deep neural network model for deciphering the adsorption of 5-Fluorouracil in COFs†
A database of 1242 experimentally synthesized COFs has been studied to understand their potential as drug carriers by employing molecular simulations and machine learning models to analyze the adsorption abilities and predict the capacity of loading the anticancer drug, 5-fluorouracil. Our findings indicate that different organic linkers, structural features, binding sites, topologies, etc. of COFs play an important role in determining the maximum loading capacity and release parameters of 5-FU. The implementation of molecular simulations-based machine learning methods for drug adsorption studies in COFs is rare in the literature. Once the model was validated, we studied the maximum loading capacity of 5-FU in a series of COFs, 102–108 and 112, from the COF database, as these exhibited a gradual trend in textural properties, aiming to understand this trend and the correlation between their structure and loading capacity. Then, we proceeded to study the adsorption process in detail in 4 of the COFs: three 2D COFs—COF-206, i.e., DCuPc–ANDI-COF; COF-362, i.e., PI-COF-3; and COF-398, i.e., Py-DBA-COF-1—and one 3D COF—COF-363, i.e., PI-COF-4. Radial distribution function and adsorption energy analyses revealed some important interactions and thermodynamic parameters leading to strong binding and slow release of 5-FU. The adsorption energy values in the top-performing COFs fall within the range of −8.43 to −42.25 × 103 kJ mol−1. The correlation of ML input parameters in terms of various chemical and structural descriptors with the maximum loading capacity is discussed. From the molecular simulations, COF-362 is the best-performing COF in terms of loading capacity and adsorption energy values. The ML models, i.e., random forest, decision tree and three deep neural networks, were trained on 80% of the total data, while the remaining 20% of the data was used to test the models. DNN model-3 was chosen as the final model for further analysis based on R2 = 0.87, RMSE = 189.81, and MAE = 100.87. SHapley Additive exPlanations (SHAP) analysis and the feature importance chart indicated that among the structural descriptors, Sacc, LCD, and Vf, and among the chemical descriptors, C, H, and N, had the most positive impact on the output predictions of the model. Finally, a graphical user interface based on the best-performing ML model was created to predict the 5-FU loading capacity of COFs. This will save users time without the need to run the code or perform various tedious drug-loading experiments.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


