利用基于捕获的宏基因组评估污水和贝类样本中人类肠道病毒的多样性
IF 4.9
3区 环境科学与生态学
Q1 ENVIRONMENTAL SCIENCES
引用次数: 0
摘要
人类污水是人类肠道病毒污染环境水体的主要来源,可污染贝类等食物。宏基因组是通过先验的大规模平行测序方法分析病毒多样性的一种新方法。然而,由于污水或贝类基质中病毒载量低、病毒属多样性大以及大量基质掩盖病毒序列,对肠道病毒的精确鉴定仍然具有挑战性。这项工作比较了在文库制备过程中使用基于捕获的富集的三种商业试剂盒,以检测到的肠道病毒的多样性以及污水和贝类样品中病毒菌株的鉴定,重点关注影响人类健康的四个家族。每个样品和试剂盒准备三份文库。所有三种试剂盒都可以表征各种病毒属。在污水样品中,获得了大量长的contigs,从而可以精确鉴定超过35种菌株。在贝类样本中,长序列较少,但可以鉴定出一种人类星状病毒和一种诺如病毒株。在测试的试剂盒中,一个在重复之间显示出较低的差异,允许对来自四个感兴趣的家族的更高多样性的病毒进行测序,并产生更多的几乎完整的基因组。本文章由计算机程序翻译,如有差异,请以英文原文为准。
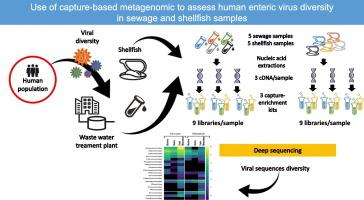
Use of capture based metagenomic to assess human enteric virus diversity in sewage and shellfish samples
Human sewage is the main source of contamination of environmental waters with human enteric viruses, that can contaminate food such as shellfish. Metagenomic represents a new way of analyzing viral diversity through an a priori massive parallel sequencing approach. However, the precise identification of enteric viruses in sewage or shellfish matrices, is still challenging due to the low viral load, large diversity of viral genera and the large amounts of matrix masking viral sequences. This work compared three commercial kits using capture-based enrichment during the library preparation, for the diversity of detected enteric viruses and for the identification of viral strains in sewage and shellfish samples, focusing on four families impacting human health. Triplicate libraries were prepared for each sample and each kit. All three kits allowed the characterization of a variety of viral genera. In sewage samples, a large number of long contigs was obtained allowing a precise identification of more than 35 strains. In shellfish samples, long contigs were rarer but allowed the identification of one human astrovirus and one norovirus strains. Of the tested kits, one displayed lower variation between replicates, allowed to sequence a higher diversity of viruses from the four families of interest and yielded a higher number of nearly-whole genomes.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Marine pollution bulletin
环境科学-海洋与淡水生物学
CiteScore
10.20
自引率
15.50%
发文量
1077
审稿时长
68 days
期刊介绍:
Marine Pollution Bulletin is concerned with the rational use of maritime and marine resources in estuaries, the seas and oceans, as well as with documenting marine pollution and introducing new forms of measurement and analysis. A wide range of topics are discussed as news, comment, reviews and research reports, not only on effluent disposal and pollution control, but also on the management, economic aspects and protection of the marine environment in general.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


