Chenyang Wei, Wenbo Mu, Hongyuan Zhang, Zhenghui Liu and Tiancheng Mu
{"title":"数据导向设计双原子催化剂提高电催化性能","authors":"Chenyang Wei, Wenbo Mu, Hongyuan Zhang, Zhenghui Liu and Tiancheng Mu","doi":"10.1039/D5TA03021H","DOIUrl":null,"url":null,"abstract":"<p >Double-atom catalysts (DACs) are promising electrocatalysts due to their synergistic metal–metal interactions and high atom utilization. However, the vast chemical space arising from diverse metal pairs and substrates presents a major challenge for rational design. Here, we combine high-throughput density functional theory (DFT) calculations with machine learning (ML) analysis to systematically investigate DACs for the CO<small><sub>2</sub></small> reduction reaction (CO<small><sub>2</sub></small>RR), hydrogen evolution reaction (HER), and oxygen evolution reaction (OER). We establish a predictive ML framework capable of rapidly screening DAC candidates with near-DFT accuracy, enabling efficient evaluation across a wide range of substrates. Guided by ML and DFT approaches, we identify PtZn/N-C<small><sub>3</sub></small>N<small><sub>4</sub></small> as a highly active OER catalyst with a theoretical overpotential of ∼0.15 eV, and CuNi/N-C<small><sub>3</sub></small>N<small><sub>4</sub></small> as a top-performing bifunctional catalyst for overall water splitting. For CO<small><sub>2</sub></small>RR, VTi/N-C<small><sub>3</sub></small>N<small><sub>4</sub></small> shows a limiting potential approaching ∼0.15 V, close to the optimal volcano plot peak, along with strong HER suppression. In summary, this work offers key insights for the design of ACs, providing substantial time savings and demonstrating the immense potential of ML as a universally applicable tool in diverse energy-related fields.</p>","PeriodicalId":82,"journal":{"name":"Journal of Materials Chemistry A","volume":" 32","pages":" 26509-26520"},"PeriodicalIF":9.5000,"publicationDate":"2025-07-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Data-guided design of double-atom catalysts for enhanced electrocatalytic performance†\",\"authors\":\"Chenyang Wei, Wenbo Mu, Hongyuan Zhang, Zhenghui Liu and Tiancheng Mu\",\"doi\":\"10.1039/D5TA03021H\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Double-atom catalysts (DACs) are promising electrocatalysts due to their synergistic metal–metal interactions and high atom utilization. However, the vast chemical space arising from diverse metal pairs and substrates presents a major challenge for rational design. Here, we combine high-throughput density functional theory (DFT) calculations with machine learning (ML) analysis to systematically investigate DACs for the CO<small><sub>2</sub></small> reduction reaction (CO<small><sub>2</sub></small>RR), hydrogen evolution reaction (HER), and oxygen evolution reaction (OER). We establish a predictive ML framework capable of rapidly screening DAC candidates with near-DFT accuracy, enabling efficient evaluation across a wide range of substrates. Guided by ML and DFT approaches, we identify PtZn/N-C<small><sub>3</sub></small>N<small><sub>4</sub></small> as a highly active OER catalyst with a theoretical overpotential of ∼0.15 eV, and CuNi/N-C<small><sub>3</sub></small>N<small><sub>4</sub></small> as a top-performing bifunctional catalyst for overall water splitting. For CO<small><sub>2</sub></small>RR, VTi/N-C<small><sub>3</sub></small>N<small><sub>4</sub></small> shows a limiting potential approaching ∼0.15 V, close to the optimal volcano plot peak, along with strong HER suppression. In summary, this work offers key insights for the design of ACs, providing substantial time savings and demonstrating the immense potential of ML as a universally applicable tool in diverse energy-related fields.</p>\",\"PeriodicalId\":82,\"journal\":{\"name\":\"Journal of Materials Chemistry A\",\"volume\":\" 32\",\"pages\":\" 26509-26520\"},\"PeriodicalIF\":9.5000,\"publicationDate\":\"2025-07-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Materials Chemistry A\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/ta/d5ta03021h\",\"RegionNum\":2,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Materials Chemistry A","FirstCategoryId":"88","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/ta/d5ta03021h","RegionNum":2,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Data-guided design of double-atom catalysts for enhanced electrocatalytic performance†
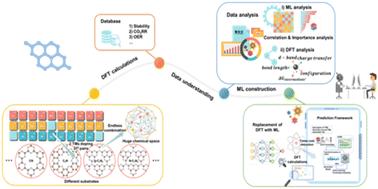
Double-atom catalysts (DACs) are promising electrocatalysts due to their synergistic metal–metal interactions and high atom utilization. However, the vast chemical space arising from diverse metal pairs and substrates presents a major challenge for rational design. Here, we combine high-throughput density functional theory (DFT) calculations with machine learning (ML) analysis to systematically investigate DACs for the CO2 reduction reaction (CO2RR), hydrogen evolution reaction (HER), and oxygen evolution reaction (OER). We establish a predictive ML framework capable of rapidly screening DAC candidates with near-DFT accuracy, enabling efficient evaluation across a wide range of substrates. Guided by ML and DFT approaches, we identify PtZn/N-C3N4 as a highly active OER catalyst with a theoretical overpotential of ∼0.15 eV, and CuNi/N-C3N4 as a top-performing bifunctional catalyst for overall water splitting. For CO2RR, VTi/N-C3N4 shows a limiting potential approaching ∼0.15 V, close to the optimal volcano plot peak, along with strong HER suppression. In summary, this work offers key insights for the design of ACs, providing substantial time savings and demonstrating the immense potential of ML as a universally applicable tool in diverse energy-related fields.
期刊介绍:
The Journal of Materials Chemistry A, B & C covers a wide range of high-quality studies in the field of materials chemistry, with each section focusing on specific applications of the materials studied. Journal of Materials Chemistry A emphasizes applications in energy and sustainability, including topics such as artificial photosynthesis, batteries, and fuel cells. Journal of Materials Chemistry B focuses on applications in biology and medicine, while Journal of Materials Chemistry C covers applications in optical, magnetic, and electronic devices. Example topic areas within the scope of Journal of Materials Chemistry A include catalysis, green/sustainable materials, sensors, and water treatment, among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


