{"title":"心脏特异性的PRMT5过表达会加剧压力超载引起的肥厚和心力衰竭。","authors":"Yasufumi Katanasaka, Yoichi Sunagawa, Ryoga Sakurai, Mikuto Tojima, Ryuya Naruta, Yuya Hojo, Yuto Kawase, Toshihide Hamabe-Horiike, Kiyoshi Mori, Koji Hasegawa, Tatsuya Morimoto","doi":"10.1186/s12929-025-01162-6","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Various epigenetic modifiers are involved in the regulation of gene expression during pathological cardiac hypertrophy-a critical event in the development of heart failure. Our previous research has demonstrated that protein arginine methyltransferase 5 (PRMT5) in cardiac fibroblasts is a crucial epigenetic writer implicated in pathological cardiac fibrosis. Moreover, treatment with a PRMT5 inhibitor also suppressed cardiac hypertrophy in mice after transverse aortic constriction (TAC) surgery. However, as the functional role of PRMT5 in cardiomyocytes remains to be fully elucidated in pathological cardiac hypertrophy and systolic dysfunction, this study aimed to clarify the gain-of-function of PRMT5 in cardiomyocytes.</p><p><strong>Methods: </strong>Cardiac-specific PRMT5 transgenic (PRMT5-TG) mice were generated to evaluate the gain-of-function of PRMT5 in cardiac hypertrophy and dysfunction in male mice undergoing TAC surgery. Cardiac function and myocardial cell hypertrophy were evaluated in wild-type (WT) and PRMT5-TG mice after TAC surgery. To elucidate the molecular mechanistic basis through which PRMT5 induces cardiomyocyte hypertrophy, we examined epigenetic modifications of histones in cardiomyocytes.</p><p><strong>Results: </strong>Echocardiography revealed that fractional shortening was reduced in PRMT5-TG mice compared to WT mice after TAC surgery. Both heart weight/BW and lung weight/BW ratios increased significantly more in PRMT5-TG than in WT mice. Histological analyses showed that cardiomyocyte diameter and perivascular fibrosis were elevated in PRMT5-TG mice in comparison to WT mice. Hypertrophic gene expression significantly increased in PRMT5-TG mice after TAC surgery. In primary cultured neonatal rat cardiac myocytes, EPZ015666, a specific inhibitor of PRMT5, and PRMT5 knockdown significantly inhibited phenylephrine (PE)-induced cell hypertrophy. Cardiac overexpression of PRMT5 promoted the acetylation of H3K9, a histone marker associated with cardiomyocyte hypertrophy, and the histone acetyltransferase activity of p300. Conversely, treatment with EPZ015666 reduced the acetylation of H3K9 induced by TAC surgery and PE treatment. Finally, we found that PRMT5 interacts with and methylates p300 at R200. The R200 point mutation in p300 abolished PRMT5-mediated enhancement of its histone acetyltransferase activity.</p><p><strong>Conclusions: </strong>The gain-of-function of PRMT5 in cardiomyocytes exacerbates pressure overload-induced cardiac hypertrophy and left ventricular systolic dysfunction, at least partially, through p300 methylation and histone acetyltransferase activation.</p>","PeriodicalId":15365,"journal":{"name":"Journal of Biomedical Science","volume":"32 1","pages":"61"},"PeriodicalIF":12.1000,"publicationDate":"2025-07-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12229037/pdf/","citationCount":"0","resultStr":"{\"title\":\"Cardiac-specific overexpression of PRMT5 exacerbates pressure overload-induced hypertrophy and heart failure.\",\"authors\":\"Yasufumi Katanasaka, Yoichi Sunagawa, Ryoga Sakurai, Mikuto Tojima, Ryuya Naruta, Yuya Hojo, Yuto Kawase, Toshihide Hamabe-Horiike, Kiyoshi Mori, Koji Hasegawa, Tatsuya Morimoto\",\"doi\":\"10.1186/s12929-025-01162-6\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Various epigenetic modifiers are involved in the regulation of gene expression during pathological cardiac hypertrophy-a critical event in the development of heart failure. Our previous research has demonstrated that protein arginine methyltransferase 5 (PRMT5) in cardiac fibroblasts is a crucial epigenetic writer implicated in pathological cardiac fibrosis. Moreover, treatment with a PRMT5 inhibitor also suppressed cardiac hypertrophy in mice after transverse aortic constriction (TAC) surgery. However, as the functional role of PRMT5 in cardiomyocytes remains to be fully elucidated in pathological cardiac hypertrophy and systolic dysfunction, this study aimed to clarify the gain-of-function of PRMT5 in cardiomyocytes.</p><p><strong>Methods: </strong>Cardiac-specific PRMT5 transgenic (PRMT5-TG) mice were generated to evaluate the gain-of-function of PRMT5 in cardiac hypertrophy and dysfunction in male mice undergoing TAC surgery. Cardiac function and myocardial cell hypertrophy were evaluated in wild-type (WT) and PRMT5-TG mice after TAC surgery. To elucidate the molecular mechanistic basis through which PRMT5 induces cardiomyocyte hypertrophy, we examined epigenetic modifications of histones in cardiomyocytes.</p><p><strong>Results: </strong>Echocardiography revealed that fractional shortening was reduced in PRMT5-TG mice compared to WT mice after TAC surgery. Both heart weight/BW and lung weight/BW ratios increased significantly more in PRMT5-TG than in WT mice. Histological analyses showed that cardiomyocyte diameter and perivascular fibrosis were elevated in PRMT5-TG mice in comparison to WT mice. Hypertrophic gene expression significantly increased in PRMT5-TG mice after TAC surgery. In primary cultured neonatal rat cardiac myocytes, EPZ015666, a specific inhibitor of PRMT5, and PRMT5 knockdown significantly inhibited phenylephrine (PE)-induced cell hypertrophy. Cardiac overexpression of PRMT5 promoted the acetylation of H3K9, a histone marker associated with cardiomyocyte hypertrophy, and the histone acetyltransferase activity of p300. Conversely, treatment with EPZ015666 reduced the acetylation of H3K9 induced by TAC surgery and PE treatment. Finally, we found that PRMT5 interacts with and methylates p300 at R200. The R200 point mutation in p300 abolished PRMT5-mediated enhancement of its histone acetyltransferase activity.</p><p><strong>Conclusions: </strong>The gain-of-function of PRMT5 in cardiomyocytes exacerbates pressure overload-induced cardiac hypertrophy and left ventricular systolic dysfunction, at least partially, through p300 methylation and histone acetyltransferase activation.</p>\",\"PeriodicalId\":15365,\"journal\":{\"name\":\"Journal of Biomedical Science\",\"volume\":\"32 1\",\"pages\":\"61\"},\"PeriodicalIF\":12.1000,\"publicationDate\":\"2025-07-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12229037/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Biomedical Science\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s12929-025-01162-6\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CELL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Biomedical Science","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12929-025-01162-6","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
Cardiac-specific overexpression of PRMT5 exacerbates pressure overload-induced hypertrophy and heart failure.
Background: Various epigenetic modifiers are involved in the regulation of gene expression during pathological cardiac hypertrophy-a critical event in the development of heart failure. Our previous research has demonstrated that protein arginine methyltransferase 5 (PRMT5) in cardiac fibroblasts is a crucial epigenetic writer implicated in pathological cardiac fibrosis. Moreover, treatment with a PRMT5 inhibitor also suppressed cardiac hypertrophy in mice after transverse aortic constriction (TAC) surgery. However, as the functional role of PRMT5 in cardiomyocytes remains to be fully elucidated in pathological cardiac hypertrophy and systolic dysfunction, this study aimed to clarify the gain-of-function of PRMT5 in cardiomyocytes.
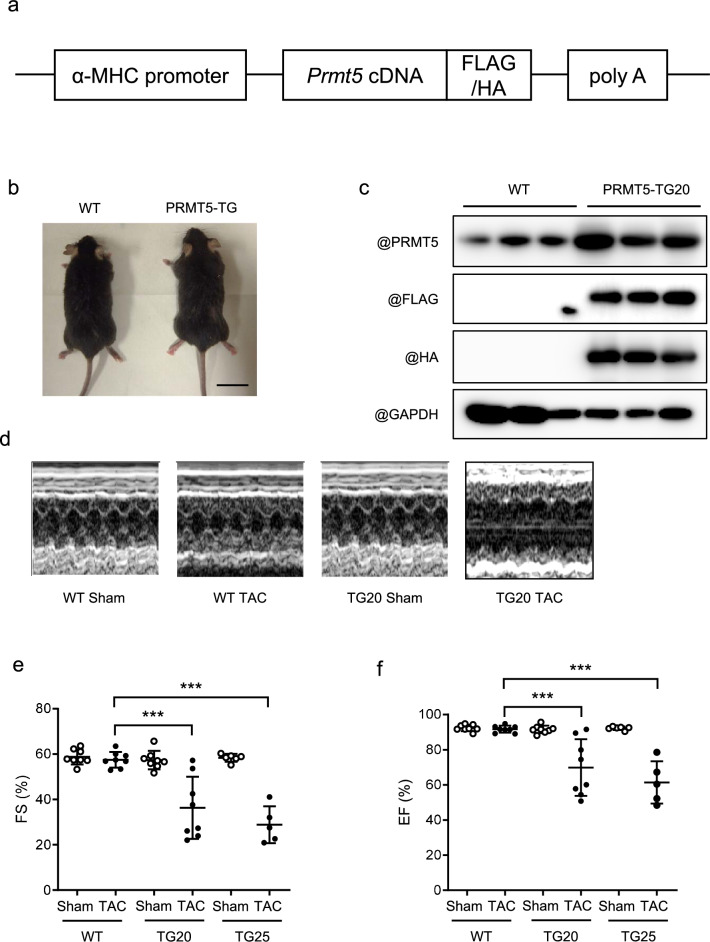
Methods: Cardiac-specific PRMT5 transgenic (PRMT5-TG) mice were generated to evaluate the gain-of-function of PRMT5 in cardiac hypertrophy and dysfunction in male mice undergoing TAC surgery. Cardiac function and myocardial cell hypertrophy were evaluated in wild-type (WT) and PRMT5-TG mice after TAC surgery. To elucidate the molecular mechanistic basis through which PRMT5 induces cardiomyocyte hypertrophy, we examined epigenetic modifications of histones in cardiomyocytes.
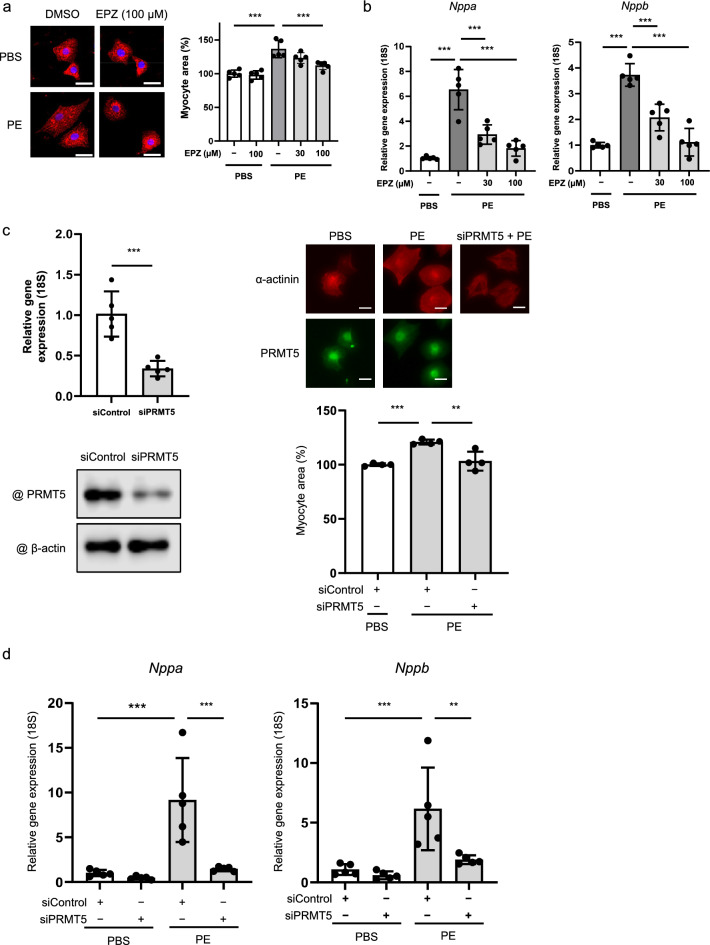
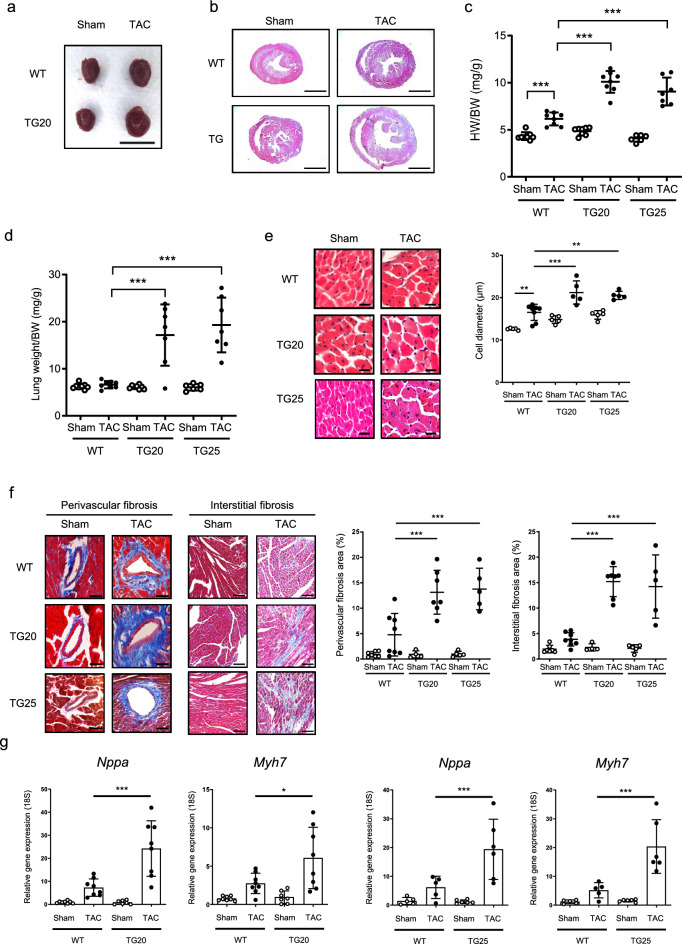
Results: Echocardiography revealed that fractional shortening was reduced in PRMT5-TG mice compared to WT mice after TAC surgery. Both heart weight/BW and lung weight/BW ratios increased significantly more in PRMT5-TG than in WT mice. Histological analyses showed that cardiomyocyte diameter and perivascular fibrosis were elevated in PRMT5-TG mice in comparison to WT mice. Hypertrophic gene expression significantly increased in PRMT5-TG mice after TAC surgery. In primary cultured neonatal rat cardiac myocytes, EPZ015666, a specific inhibitor of PRMT5, and PRMT5 knockdown significantly inhibited phenylephrine (PE)-induced cell hypertrophy. Cardiac overexpression of PRMT5 promoted the acetylation of H3K9, a histone marker associated with cardiomyocyte hypertrophy, and the histone acetyltransferase activity of p300. Conversely, treatment with EPZ015666 reduced the acetylation of H3K9 induced by TAC surgery and PE treatment. Finally, we found that PRMT5 interacts with and methylates p300 at R200. The R200 point mutation in p300 abolished PRMT5-mediated enhancement of its histone acetyltransferase activity.
Conclusions: The gain-of-function of PRMT5 in cardiomyocytes exacerbates pressure overload-induced cardiac hypertrophy and left ventricular systolic dysfunction, at least partially, through p300 methylation and histone acetyltransferase activation.
期刊介绍:
The Journal of Biomedical Science is an open access, peer-reviewed journal that focuses on fundamental and molecular aspects of basic medical sciences. It emphasizes molecular studies of biomedical problems and mechanisms. The National Science and Technology Council (NSTC), Taiwan supports the journal and covers the publication costs for accepted articles. The journal aims to provide an international platform for interdisciplinary discussions and contribute to the advancement of medicine. It benefits both readers and authors by accelerating the dissemination of research information and providing maximum access to scholarly communication. All articles published in the Journal of Biomedical Science are included in various databases such as Biological Abstracts, BIOSIS, CABI, CAS, Citebase, Current contents, DOAJ, Embase, EmBiology, and Global Health, among others.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


