Linlin Zhang, Nan Chen, Hongbo Wu, Zhen Gao, Yanning Wang and Kai Xiong
{"title":"N、O配位对co基单原子催化剂双官能团氧电催化活性影响的理论研究","authors":"Linlin Zhang, Nan Chen, Hongbo Wu, Zhen Gao, Yanning Wang and Kai Xiong","doi":"10.1039/D5NJ01527H","DOIUrl":null,"url":null,"abstract":"<p >Single-atom catalysts (SACs) have garnered considerable attention due to their outstanding electrocatalytic performance in the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER). Their key advantage lies in the ability to significantly activate the intrinsic activity of the central metal atom through fine-tuning of the coordination environment. In this study, density functional theory calculations were employed to systematically investigate a series of graphene-based cobalt SACs. By modulating the N/O coordination environment around the Co atom, the stability and bifunctional electrocatalytic performance of these catalysts were evaluated. The results show that CoN<small><sub>3</sub></small>O exhibits excellent bifunctional catalytic activity (<em>η</em><small><sup>ORR</sup></small> = 0.44 V, <em>η</em><small><sup>OER</sup></small> = 0.37 V). Furthermore, a correlation between the adsorption free energies of intermediates and the O–Co bond length (<em>d</em><small><sub>O–Co</sub></small>) was identified: the adsorption free energies of *OOH and *OH show a positive correlation with <em>d</em><small><sub>O–Co</sub></small>, while that of *O is independent of <em>d</em><small><sub>O–Co</sub></small>. Projected density of states and charge density difference analyses reveal that strong hybridization between Co 3d and O 2p orbitals, along with significant charge transfer, enhances the coupling between active sites and reactants, thereby improving catalytic activity. This study highlights the crucial role of coordination environment and electronic structure in tuning the performance of SACs.</p>","PeriodicalId":95,"journal":{"name":"New Journal of Chemistry","volume":" 27","pages":" 11667-11676"},"PeriodicalIF":2.5000,"publicationDate":"2025-06-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Theoretical investigation of N and O coordination effects on the bifunctional oxygen electrocatalytic activity of Co-based single-atom catalysts†\",\"authors\":\"Linlin Zhang, Nan Chen, Hongbo Wu, Zhen Gao, Yanning Wang and Kai Xiong\",\"doi\":\"10.1039/D5NJ01527H\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Single-atom catalysts (SACs) have garnered considerable attention due to their outstanding electrocatalytic performance in the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER). Their key advantage lies in the ability to significantly activate the intrinsic activity of the central metal atom through fine-tuning of the coordination environment. In this study, density functional theory calculations were employed to systematically investigate a series of graphene-based cobalt SACs. By modulating the N/O coordination environment around the Co atom, the stability and bifunctional electrocatalytic performance of these catalysts were evaluated. The results show that CoN<small><sub>3</sub></small>O exhibits excellent bifunctional catalytic activity (<em>η</em><small><sup>ORR</sup></small> = 0.44 V, <em>η</em><small><sup>OER</sup></small> = 0.37 V). Furthermore, a correlation between the adsorption free energies of intermediates and the O–Co bond length (<em>d</em><small><sub>O–Co</sub></small>) was identified: the adsorption free energies of *OOH and *OH show a positive correlation with <em>d</em><small><sub>O–Co</sub></small>, while that of *O is independent of <em>d</em><small><sub>O–Co</sub></small>. Projected density of states and charge density difference analyses reveal that strong hybridization between Co 3d and O 2p orbitals, along with significant charge transfer, enhances the coupling between active sites and reactants, thereby improving catalytic activity. This study highlights the crucial role of coordination environment and electronic structure in tuning the performance of SACs.</p>\",\"PeriodicalId\":95,\"journal\":{\"name\":\"New Journal of Chemistry\",\"volume\":\" 27\",\"pages\":\" 11667-11676\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2025-06-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"New Journal of Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/nj/d5nj01527h\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"New Journal of Chemistry","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/nj/d5nj01527h","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Theoretical investigation of N and O coordination effects on the bifunctional oxygen electrocatalytic activity of Co-based single-atom catalysts†
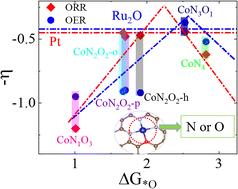
Single-atom catalysts (SACs) have garnered considerable attention due to their outstanding electrocatalytic performance in the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER). Their key advantage lies in the ability to significantly activate the intrinsic activity of the central metal atom through fine-tuning of the coordination environment. In this study, density functional theory calculations were employed to systematically investigate a series of graphene-based cobalt SACs. By modulating the N/O coordination environment around the Co atom, the stability and bifunctional electrocatalytic performance of these catalysts were evaluated. The results show that CoN3O exhibits excellent bifunctional catalytic activity (ηORR = 0.44 V, ηOER = 0.37 V). Furthermore, a correlation between the adsorption free energies of intermediates and the O–Co bond length (dO–Co) was identified: the adsorption free energies of *OOH and *OH show a positive correlation with dO–Co, while that of *O is independent of dO–Co. Projected density of states and charge density difference analyses reveal that strong hybridization between Co 3d and O 2p orbitals, along with significant charge transfer, enhances the coupling between active sites and reactants, thereby improving catalytic activity. This study highlights the crucial role of coordination environment and electronic structure in tuning the performance of SACs.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


