Paula Bruna Mattos Coelho Araujo, Luiz Filipe Rocha de Sá, Rafaela Sousa, Darine Villela, Thereza Taylanne Souza Loureiro Cavalcanti, Michele Patricia Migliavacca, Marilia Martins Guimaraes, Micheline Abreu Rayol Souza, Erika Naliato, Mariana Botelho, João Bosco Nascimento, Mirna Sanchez Carvallo, Pedro Martins Viveiros, Delmar Muniz Lourenço Junior, Rosita Fontes, Alice Helena Dutra Violante
{"title":"父子中枢性尿崩症与一罕见变异有关:1例报告。","authors":"Paula Bruna Mattos Coelho Araujo, Luiz Filipe Rocha de Sá, Rafaela Sousa, Darine Villela, Thereza Taylanne Souza Loureiro Cavalcanti, Michele Patricia Migliavacca, Marilia Martins Guimaraes, Micheline Abreu Rayol Souza, Erika Naliato, Mariana Botelho, João Bosco Nascimento, Mirna Sanchez Carvallo, Pedro Martins Viveiros, Delmar Muniz Lourenço Junior, Rosita Fontes, Alice Helena Dutra Violante","doi":"10.1159/000543795","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Central diabetes insipidus (CDI) is a rare disorder caused by a deficiency in the secretion of arginine vasopressin (AVP) from the posterior pituitary. It can be either acquired or congenital, often due to genetic factors, and is typically inherited in an autosomal dominant manner.</p><p><strong>Case presentation: </strong>This study describes the clinical features and the genetic analysis of a father and his son with familial CDI. Both presented in childhood with typical symptoms, including polyuria, polydipsia, and hypernatremia. Diagnosis was confirmed through water deprivation testing and subsequently supported by sellar magnetic resonance imaging. Genetic analysis identified the rare germline variant c.329G>A (p.Cys110Tyr) in the <i>AVP</i> gene, in heterozygosity, which segregated in the parent-child pair, thereby elucidating the familial basis of the CDI.</p><p><strong>Conclusion: </strong>This rare germline <i>AVP</i> variant causes a cysteine-to-tyrosine amino acid substitution in the encoded protein and is classified as pathogenic. Familial cases of rare diseases, such as CDI, highlight the importance of clinical evaluation of relatives with similar symptoms and emphasize the need for molecular studies and genetic counseling.</p>","PeriodicalId":101351,"journal":{"name":"Biomedicine hub","volume":"10 1","pages":"57-63"},"PeriodicalIF":0.0000,"publicationDate":"2025-02-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12215092/pdf/","citationCount":"0","resultStr":"{\"title\":\"Central Diabetes Insipidus in Father and Son Linked to a Rare Variant: A Case Report.\",\"authors\":\"Paula Bruna Mattos Coelho Araujo, Luiz Filipe Rocha de Sá, Rafaela Sousa, Darine Villela, Thereza Taylanne Souza Loureiro Cavalcanti, Michele Patricia Migliavacca, Marilia Martins Guimaraes, Micheline Abreu Rayol Souza, Erika Naliato, Mariana Botelho, João Bosco Nascimento, Mirna Sanchez Carvallo, Pedro Martins Viveiros, Delmar Muniz Lourenço Junior, Rosita Fontes, Alice Helena Dutra Violante\",\"doi\":\"10.1159/000543795\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Introduction: </strong>Central diabetes insipidus (CDI) is a rare disorder caused by a deficiency in the secretion of arginine vasopressin (AVP) from the posterior pituitary. It can be either acquired or congenital, often due to genetic factors, and is typically inherited in an autosomal dominant manner.</p><p><strong>Case presentation: </strong>This study describes the clinical features and the genetic analysis of a father and his son with familial CDI. Both presented in childhood with typical symptoms, including polyuria, polydipsia, and hypernatremia. Diagnosis was confirmed through water deprivation testing and subsequently supported by sellar magnetic resonance imaging. Genetic analysis identified the rare germline variant c.329G>A (p.Cys110Tyr) in the <i>AVP</i> gene, in heterozygosity, which segregated in the parent-child pair, thereby elucidating the familial basis of the CDI.</p><p><strong>Conclusion: </strong>This rare germline <i>AVP</i> variant causes a cysteine-to-tyrosine amino acid substitution in the encoded protein and is classified as pathogenic. Familial cases of rare diseases, such as CDI, highlight the importance of clinical evaluation of relatives with similar symptoms and emphasize the need for molecular studies and genetic counseling.</p>\",\"PeriodicalId\":101351,\"journal\":{\"name\":\"Biomedicine hub\",\"volume\":\"10 1\",\"pages\":\"57-63\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2025-02-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12215092/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biomedicine hub\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1159/000543795\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biomedicine hub","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1159/000543795","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Central Diabetes Insipidus in Father and Son Linked to a Rare Variant: A Case Report.
Introduction: Central diabetes insipidus (CDI) is a rare disorder caused by a deficiency in the secretion of arginine vasopressin (AVP) from the posterior pituitary. It can be either acquired or congenital, often due to genetic factors, and is typically inherited in an autosomal dominant manner.
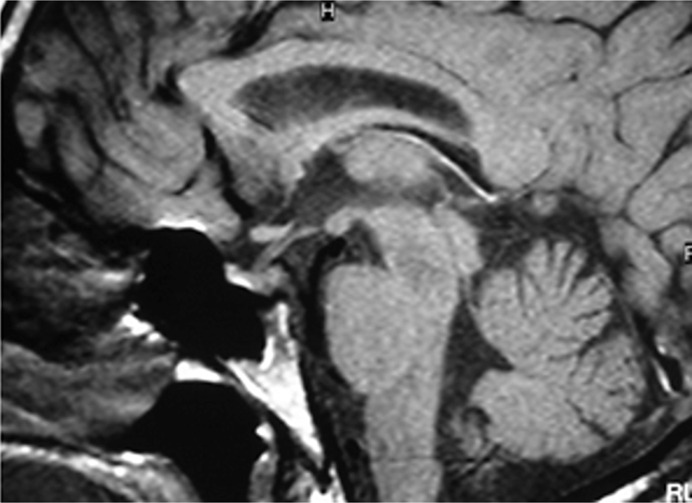
Case presentation: This study describes the clinical features and the genetic analysis of a father and his son with familial CDI. Both presented in childhood with typical symptoms, including polyuria, polydipsia, and hypernatremia. Diagnosis was confirmed through water deprivation testing and subsequently supported by sellar magnetic resonance imaging. Genetic analysis identified the rare germline variant c.329G>A (p.Cys110Tyr) in the AVP gene, in heterozygosity, which segregated in the parent-child pair, thereby elucidating the familial basis of the CDI.
Conclusion: This rare germline AVP variant causes a cysteine-to-tyrosine amino acid substitution in the encoded protein and is classified as pathogenic. Familial cases of rare diseases, such as CDI, highlight the importance of clinical evaluation of relatives with similar symptoms and emphasize the need for molecular studies and genetic counseling.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


