Shiva Kazempour Dehkordi, Sogand Sajedi, Amirreza Heshmat, Miranda E Orr, Habil Zare
{"title":"通过对死后人类大脑的转录组学分析鉴定神经新生标志物。","authors":"Shiva Kazempour Dehkordi, Sogand Sajedi, Amirreza Heshmat, Miranda E Orr, Habil Zare","doi":"10.1038/s41514-025-00235-y","DOIUrl":null,"url":null,"abstract":"<p><p>Neuronal senescence (i.e., neurescence) is an important hallmark of aging and neurodegeneration, but it remains poorly characterized in the human brain due to the lack of reliable markers. This study aimed to identify neurescence markers based on single-nucleus transcriptome data from postmortem human prefrontal cortex. Using an eigengene approach, we integrated three gene panels: (a) SenMayo, (b) canonical senescence pathway (CSP), and (c) senescence initiating pathway (SIP), to identify neurescence signatures. We found that paired markers outperform single markers; for instance, by combining CDKN2D and ETS2 in a decision tree, a high accuracy of 99% and perfect specificity (100%) were achieved in distinguishing senescent neurons (i.e, neurescent). Differential expression analyses identified 324 genes that are overexpressed in neurescent. These genes showed significant associations with important neurodegeneration-related pathways, including Alzheimer's disease, Parkinson's disease, and Huntington's disease. Interestingly, several of these overexpressed genes are linked to mitochondrial dysfunction and cytoskeletal dysregulation. These findings provide valuable insights into the complexities of neurescence, emphasizing the need for further exploration of histologically viable markers and validation in broader datasets.</p>","PeriodicalId":94160,"journal":{"name":"npj aging","volume":"11 1","pages":"57"},"PeriodicalIF":6.0000,"publicationDate":"2025-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12214559/pdf/","citationCount":"0","resultStr":"{\"title\":\"Identification of markers for neurescence through transcriptomic profiling of postmortem human brains.\",\"authors\":\"Shiva Kazempour Dehkordi, Sogand Sajedi, Amirreza Heshmat, Miranda E Orr, Habil Zare\",\"doi\":\"10.1038/s41514-025-00235-y\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Neuronal senescence (i.e., neurescence) is an important hallmark of aging and neurodegeneration, but it remains poorly characterized in the human brain due to the lack of reliable markers. This study aimed to identify neurescence markers based on single-nucleus transcriptome data from postmortem human prefrontal cortex. Using an eigengene approach, we integrated three gene panels: (a) SenMayo, (b) canonical senescence pathway (CSP), and (c) senescence initiating pathway (SIP), to identify neurescence signatures. We found that paired markers outperform single markers; for instance, by combining CDKN2D and ETS2 in a decision tree, a high accuracy of 99% and perfect specificity (100%) were achieved in distinguishing senescent neurons (i.e, neurescent). Differential expression analyses identified 324 genes that are overexpressed in neurescent. These genes showed significant associations with important neurodegeneration-related pathways, including Alzheimer's disease, Parkinson's disease, and Huntington's disease. Interestingly, several of these overexpressed genes are linked to mitochondrial dysfunction and cytoskeletal dysregulation. These findings provide valuable insights into the complexities of neurescence, emphasizing the need for further exploration of histologically viable markers and validation in broader datasets.</p>\",\"PeriodicalId\":94160,\"journal\":{\"name\":\"npj aging\",\"volume\":\"11 1\",\"pages\":\"57\"},\"PeriodicalIF\":6.0000,\"publicationDate\":\"2025-07-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12214559/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"npj aging\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1038/s41514-025-00235-y\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GERIATRICS & GERONTOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj aging","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1038/s41514-025-00235-y","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GERIATRICS & GERONTOLOGY","Score":null,"Total":0}
Identification of markers for neurescence through transcriptomic profiling of postmortem human brains.
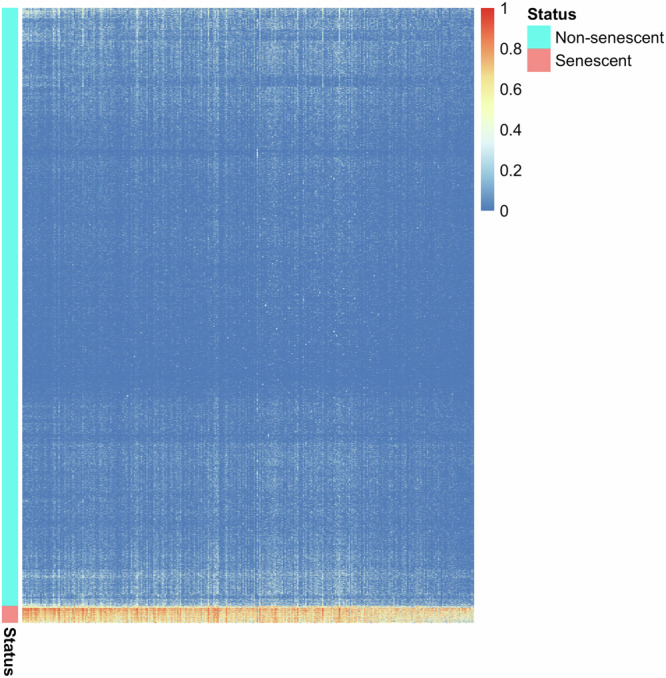
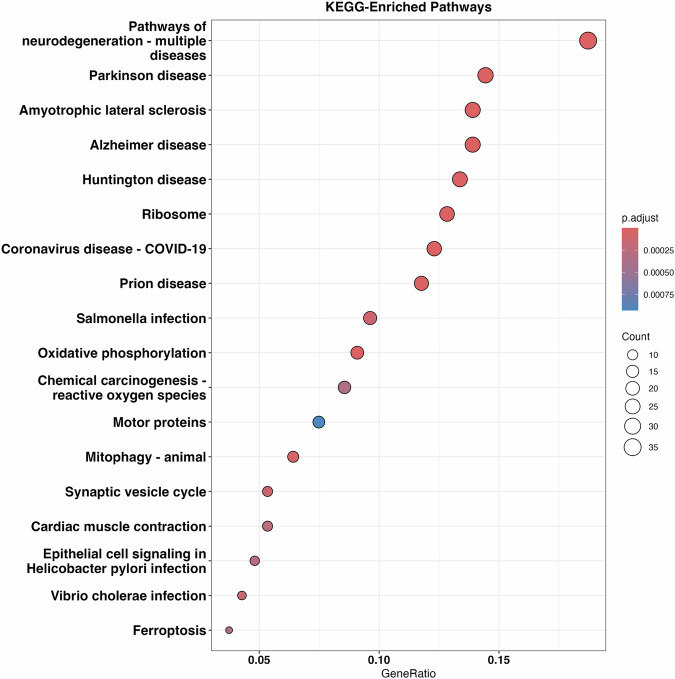
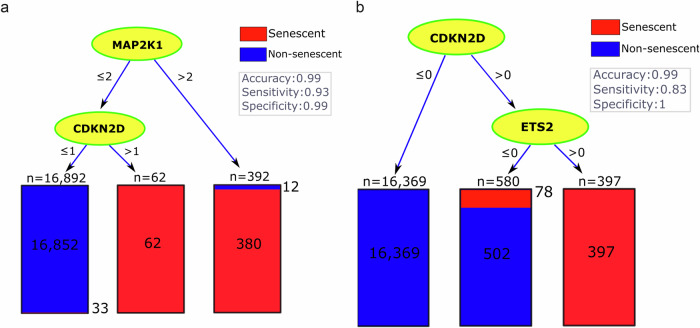
Neuronal senescence (i.e., neurescence) is an important hallmark of aging and neurodegeneration, but it remains poorly characterized in the human brain due to the lack of reliable markers. This study aimed to identify neurescence markers based on single-nucleus transcriptome data from postmortem human prefrontal cortex. Using an eigengene approach, we integrated three gene panels: (a) SenMayo, (b) canonical senescence pathway (CSP), and (c) senescence initiating pathway (SIP), to identify neurescence signatures. We found that paired markers outperform single markers; for instance, by combining CDKN2D and ETS2 in a decision tree, a high accuracy of 99% and perfect specificity (100%) were achieved in distinguishing senescent neurons (i.e, neurescent). Differential expression analyses identified 324 genes that are overexpressed in neurescent. These genes showed significant associations with important neurodegeneration-related pathways, including Alzheimer's disease, Parkinson's disease, and Huntington's disease. Interestingly, several of these overexpressed genes are linked to mitochondrial dysfunction and cytoskeletal dysregulation. These findings provide valuable insights into the complexities of neurescence, emphasizing the need for further exploration of histologically viable markers and validation in broader datasets.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


