Emily Kutrieb, Montserrat Vera Llonch, Derek Weycker, Steven M Kymes, Duncan Brown, Anne V Smith, Robert S Pulido, Brian Appleby
{"title":"美国克雅氏病患者的诊断过程和医疗负担:一项真实世界的证据研究","authors":"Emily Kutrieb, Montserrat Vera Llonch, Derek Weycker, Steven M Kymes, Duncan Brown, Anne V Smith, Robert S Pulido, Brian Appleby","doi":"10.1212/CPJ.0000000000200502","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and objectives: </strong>Evidence on the diagnostic journey and health care burden of patients with Creutzfeldt-Jakob disease (CJD) in the United States is limited. A real-world evidence study using a US health care claims database was undertaken to address this gap.</p><p><strong>Methods: </strong>A retrospective observational cohort study was conducted using data from the Merative MarketScan Research Databases (01/2012-12/2020). Study population comprised adults aged 18 years or older with evidence of CJD (initial diagnosis = index date), no evidence of selected neurologic conditions after the last CJD diagnosis, and health care coverage during the 12-month pre-index period; adults meeting selection criteria are referred herein as \"patients with CJD.\" Diagnostic journey was detailed based on evidence of symptoms and alternative neurologic conditions during the pre-index period as well as time to death (based on a proxy). Health care burden was summarized through levels of all-cause health care utilization and expenditures during the pre/post-index periods.</p><p><strong>Results: </strong>A total of 215 patients with CJD qualified for inclusion in the study population. The mean duration from first symptom to initial CJD diagnosis was 5.0 months, and 80% of patients had ≥3 symptoms, most commonly altered mental status (82%), gait/coordination disturbance (60%), and malaise/fatigue (44%). Most patients (63%) also had ≥1 alternative diagnosis, including cerebrovascular disease (49%), peripheral vertigo (11%), and Alzheimer disease (7%); the mean duration from first alternative diagnosis to initial CJD diagnosis was 2.4 months. The mean (median) time to death (proxy) from first symptom was 7.9 (6.6) months and from initial CJD diagnosis was 2.9 (1.1) months. During the 12-month pre-index period, mean (95% CI) cumulative health care expenditures were $35,493 ($28,914-$42,722); by the end of the post-index period, cumulative expenditures averaged $93,601 ($78,878-$109,776) per patient.</p><p><strong>Discussion: </strong>Study findings suggest that, in US clinical practice, patients with CJD present with one or more clinical symptoms affecting motor, cognitive, or other domains, and many alternative diagnoses are considered, which may prolong the diagnostic journey. Study findings also suggest that health care expenditures-especially proximate to the initial CJD diagnosis-are notably high. CJD should be considered in the differential diagnosis of adults with rapidly progressing dementia or motor disturbance.</p>","PeriodicalId":19136,"journal":{"name":"Neurology. Clinical practice","volume":"15 4","pages":"e200502"},"PeriodicalIF":3.2000,"publicationDate":"2025-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12204766/pdf/","citationCount":"0","resultStr":"{\"title\":\"Diagnostic Journey and Health Care Burden of Patients With Creutzfeldt-Jakob Disease in the United States: A Real-World Evidence Study.\",\"authors\":\"Emily Kutrieb, Montserrat Vera Llonch, Derek Weycker, Steven M Kymes, Duncan Brown, Anne V Smith, Robert S Pulido, Brian Appleby\",\"doi\":\"10.1212/CPJ.0000000000200502\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background and objectives: </strong>Evidence on the diagnostic journey and health care burden of patients with Creutzfeldt-Jakob disease (CJD) in the United States is limited. A real-world evidence study using a US health care claims database was undertaken to address this gap.</p><p><strong>Methods: </strong>A retrospective observational cohort study was conducted using data from the Merative MarketScan Research Databases (01/2012-12/2020). Study population comprised adults aged 18 years or older with evidence of CJD (initial diagnosis = index date), no evidence of selected neurologic conditions after the last CJD diagnosis, and health care coverage during the 12-month pre-index period; adults meeting selection criteria are referred herein as \\\"patients with CJD.\\\" Diagnostic journey was detailed based on evidence of symptoms and alternative neurologic conditions during the pre-index period as well as time to death (based on a proxy). Health care burden was summarized through levels of all-cause health care utilization and expenditures during the pre/post-index periods.</p><p><strong>Results: </strong>A total of 215 patients with CJD qualified for inclusion in the study population. The mean duration from first symptom to initial CJD diagnosis was 5.0 months, and 80% of patients had ≥3 symptoms, most commonly altered mental status (82%), gait/coordination disturbance (60%), and malaise/fatigue (44%). Most patients (63%) also had ≥1 alternative diagnosis, including cerebrovascular disease (49%), peripheral vertigo (11%), and Alzheimer disease (7%); the mean duration from first alternative diagnosis to initial CJD diagnosis was 2.4 months. The mean (median) time to death (proxy) from first symptom was 7.9 (6.6) months and from initial CJD diagnosis was 2.9 (1.1) months. During the 12-month pre-index period, mean (95% CI) cumulative health care expenditures were $35,493 ($28,914-$42,722); by the end of the post-index period, cumulative expenditures averaged $93,601 ($78,878-$109,776) per patient.</p><p><strong>Discussion: </strong>Study findings suggest that, in US clinical practice, patients with CJD present with one or more clinical symptoms affecting motor, cognitive, or other domains, and many alternative diagnoses are considered, which may prolong the diagnostic journey. Study findings also suggest that health care expenditures-especially proximate to the initial CJD diagnosis-are notably high. CJD should be considered in the differential diagnosis of adults with rapidly progressing dementia or motor disturbance.</p>\",\"PeriodicalId\":19136,\"journal\":{\"name\":\"Neurology. Clinical practice\",\"volume\":\"15 4\",\"pages\":\"e200502\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2025-08-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12204766/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neurology. Clinical practice\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1212/CPJ.0000000000200502\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/6/25 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology. Clinical practice","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1212/CPJ.0000000000200502","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/6/25 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Diagnostic Journey and Health Care Burden of Patients With Creutzfeldt-Jakob Disease in the United States: A Real-World Evidence Study.
Background and objectives: Evidence on the diagnostic journey and health care burden of patients with Creutzfeldt-Jakob disease (CJD) in the United States is limited. A real-world evidence study using a US health care claims database was undertaken to address this gap.
Methods: A retrospective observational cohort study was conducted using data from the Merative MarketScan Research Databases (01/2012-12/2020). Study population comprised adults aged 18 years or older with evidence of CJD (initial diagnosis = index date), no evidence of selected neurologic conditions after the last CJD diagnosis, and health care coverage during the 12-month pre-index period; adults meeting selection criteria are referred herein as "patients with CJD." Diagnostic journey was detailed based on evidence of symptoms and alternative neurologic conditions during the pre-index period as well as time to death (based on a proxy). Health care burden was summarized through levels of all-cause health care utilization and expenditures during the pre/post-index periods.
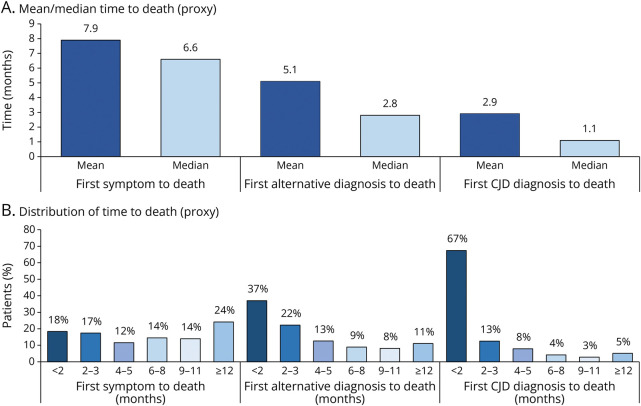
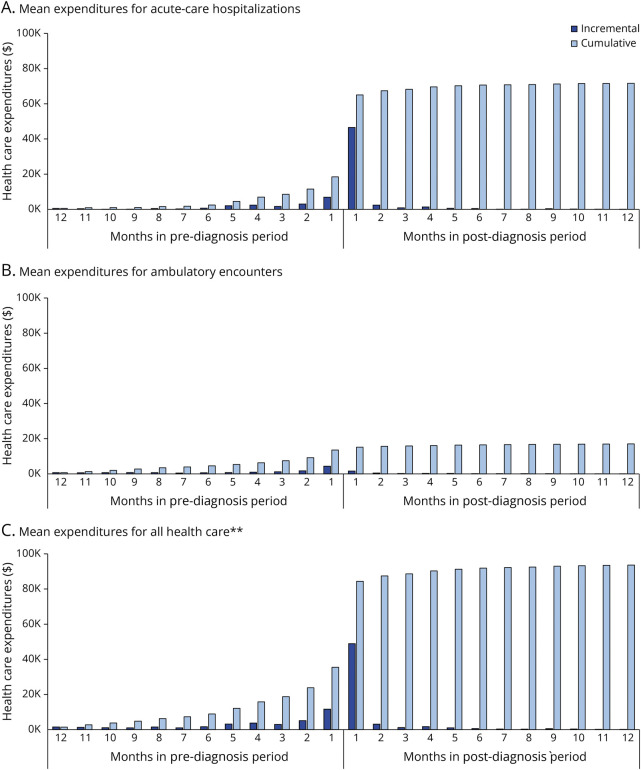
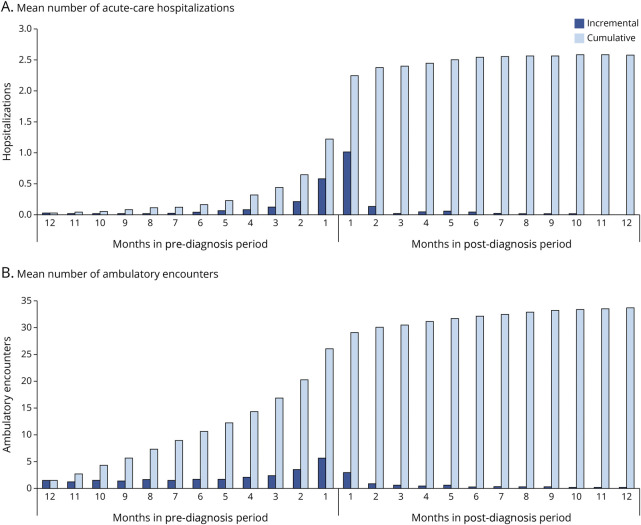
Results: A total of 215 patients with CJD qualified for inclusion in the study population. The mean duration from first symptom to initial CJD diagnosis was 5.0 months, and 80% of patients had ≥3 symptoms, most commonly altered mental status (82%), gait/coordination disturbance (60%), and malaise/fatigue (44%). Most patients (63%) also had ≥1 alternative diagnosis, including cerebrovascular disease (49%), peripheral vertigo (11%), and Alzheimer disease (7%); the mean duration from first alternative diagnosis to initial CJD diagnosis was 2.4 months. The mean (median) time to death (proxy) from first symptom was 7.9 (6.6) months and from initial CJD diagnosis was 2.9 (1.1) months. During the 12-month pre-index period, mean (95% CI) cumulative health care expenditures were $35,493 ($28,914-$42,722); by the end of the post-index period, cumulative expenditures averaged $93,601 ($78,878-$109,776) per patient.
Discussion: Study findings suggest that, in US clinical practice, patients with CJD present with one or more clinical symptoms affecting motor, cognitive, or other domains, and many alternative diagnoses are considered, which may prolong the diagnostic journey. Study findings also suggest that health care expenditures-especially proximate to the initial CJD diagnosis-are notably high. CJD should be considered in the differential diagnosis of adults with rapidly progressing dementia or motor disturbance.
期刊介绍:
Neurology® Genetics is an online open access journal publishing peer-reviewed reports in the field of neurogenetics. The journal publishes original articles in all areas of neurogenetics including rare and common genetic variations, genotype-phenotype correlations, outlier phenotypes as a result of mutations in known disease genes, and genetic variations with a putative link to diseases. Articles include studies reporting on genetic disease risk, pharmacogenomics, and results of gene-based clinical trials (viral, ASO, etc.). Genetically engineered model systems are not a primary focus of Neurology® Genetics, but studies using model systems for treatment trials, including well-powered studies reporting negative results, are welcome.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


