Lein N H Dofash, Chiara Folland, Jason Dyke, Emna Farhat, Myriam Chaabouni, Najoua Miladi, Merrilee Needham, Phillipa J Lamont, Catherine Ashton, Gianina Ravenscroft
{"title":"新的CFL2错义变异影响f -肌动蛋白解聚合并扩大CFL2相关肌病的疾病谱。","authors":"Lein N H Dofash, Chiara Folland, Jason Dyke, Emna Farhat, Myriam Chaabouni, Najoua Miladi, Merrilee Needham, Phillipa J Lamont, Catherine Ashton, Gianina Ravenscroft","doi":"10.1093/hmg/ddaf102","DOIUrl":null,"url":null,"abstract":"<p><p>Cofilin-2, encoded by CFL2, is an actin-binding protein essential for regulating actin filament dynamics in skeletal muscle. Biallelic variants in CFL2 are associated with an ultra-rare, early-onset myopathy typically presenting as nemaline myopathy. Only 10 patients have been described to date from five unrelated families. Here, we describe two new cases from two unrelated families. The first proband presented clinically with rigid spine syndrome and a biopsy keeping with nemaline myopathy. The second proband presented with a relatively mild congenital myopathy which became rapidly progressive in the fourth decade, the muscle biopsy showed cytoplasmic bodies, internal nuclei and ringbinden. Exome and genome sequencing revealed three novel biallelic missense variants in CFL2, a homozygous c.115 T > G; p.(Cys39Gly) in the proband of Family 1, and bi-allelic heterozygous c.256G > A:(p.Asp86Asn), and c.283A > G (p.Lys95Glu) variants in the proband of Family 2. We characterised the effects of these substitutions using an in vitro F-actin depolymerisation assay and showed all three were associated with significantly reduced filamentous actin depolymerisation rates compared to the wildtype. Taken together, our findings are highly suggestive of a CFL2-related disease in these patients. Since CFL2-related myopathies are ultrarare, the application of ACMG/AMP guidelines and the diagnostic reportability of CFL2 variants identified in patients remains a challenge. The actin depolymerisation assay may be useful to elucidate the impact and pathogenicity of additional CFL2 variants and has the potential to guide variant classification in future.</p>","PeriodicalId":13070,"journal":{"name":"Human molecular genetics","volume":" ","pages":"1471-1479"},"PeriodicalIF":3.2000,"publicationDate":"2025-08-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12368771/pdf/","citationCount":"0","resultStr":"{\"title\":\"Novel missense variants in CFL2 affect F-actin depolymerisation and expand the disease spectrum of CFL2-related myopathy.\",\"authors\":\"Lein N H Dofash, Chiara Folland, Jason Dyke, Emna Farhat, Myriam Chaabouni, Najoua Miladi, Merrilee Needham, Phillipa J Lamont, Catherine Ashton, Gianina Ravenscroft\",\"doi\":\"10.1093/hmg/ddaf102\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Cofilin-2, encoded by CFL2, is an actin-binding protein essential for regulating actin filament dynamics in skeletal muscle. Biallelic variants in CFL2 are associated with an ultra-rare, early-onset myopathy typically presenting as nemaline myopathy. Only 10 patients have been described to date from five unrelated families. Here, we describe two new cases from two unrelated families. The first proband presented clinically with rigid spine syndrome and a biopsy keeping with nemaline myopathy. The second proband presented with a relatively mild congenital myopathy which became rapidly progressive in the fourth decade, the muscle biopsy showed cytoplasmic bodies, internal nuclei and ringbinden. Exome and genome sequencing revealed three novel biallelic missense variants in CFL2, a homozygous c.115 T > G; p.(Cys39Gly) in the proband of Family 1, and bi-allelic heterozygous c.256G > A:(p.Asp86Asn), and c.283A > G (p.Lys95Glu) variants in the proband of Family 2. We characterised the effects of these substitutions using an in vitro F-actin depolymerisation assay and showed all three were associated with significantly reduced filamentous actin depolymerisation rates compared to the wildtype. Taken together, our findings are highly suggestive of a CFL2-related disease in these patients. Since CFL2-related myopathies are ultrarare, the application of ACMG/AMP guidelines and the diagnostic reportability of CFL2 variants identified in patients remains a challenge. The actin depolymerisation assay may be useful to elucidate the impact and pathogenicity of additional CFL2 variants and has the potential to guide variant classification in future.</p>\",\"PeriodicalId\":13070,\"journal\":{\"name\":\"Human molecular genetics\",\"volume\":\" \",\"pages\":\"1471-1479\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2025-08-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12368771/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Human molecular genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/hmg/ddaf102\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human molecular genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/hmg/ddaf102","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Novel missense variants in CFL2 affect F-actin depolymerisation and expand the disease spectrum of CFL2-related myopathy.
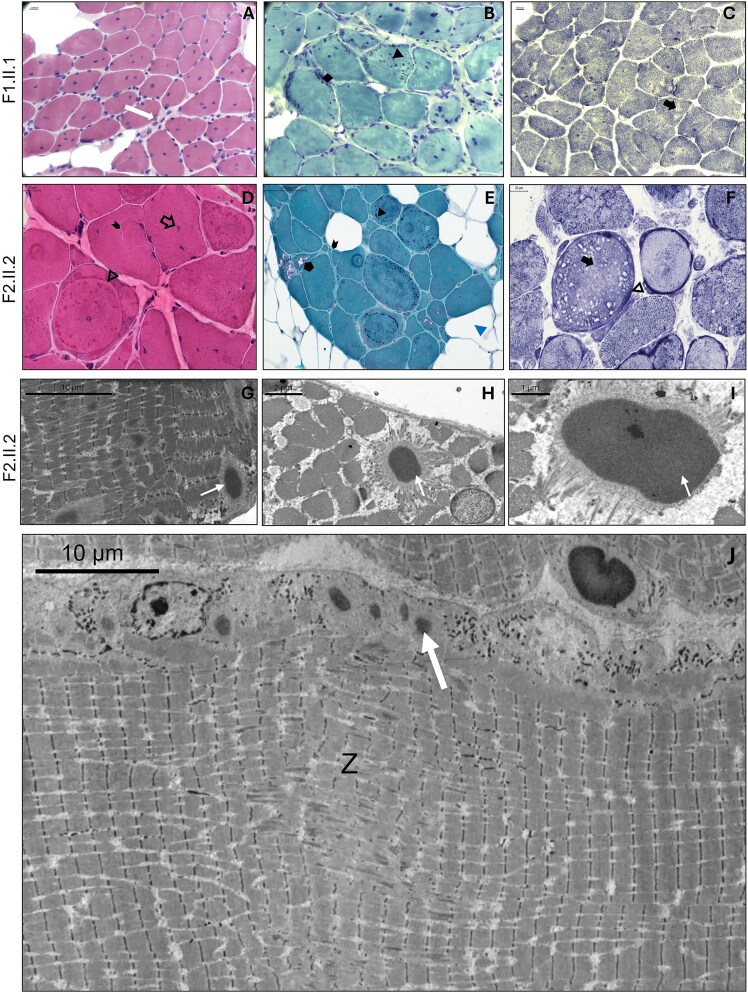
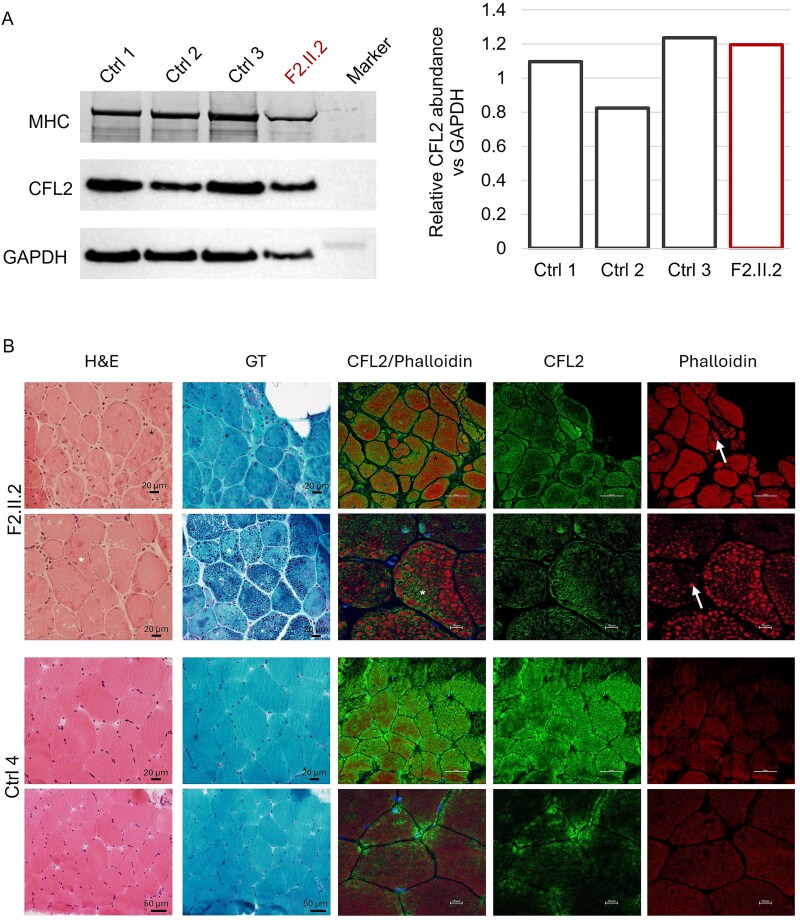
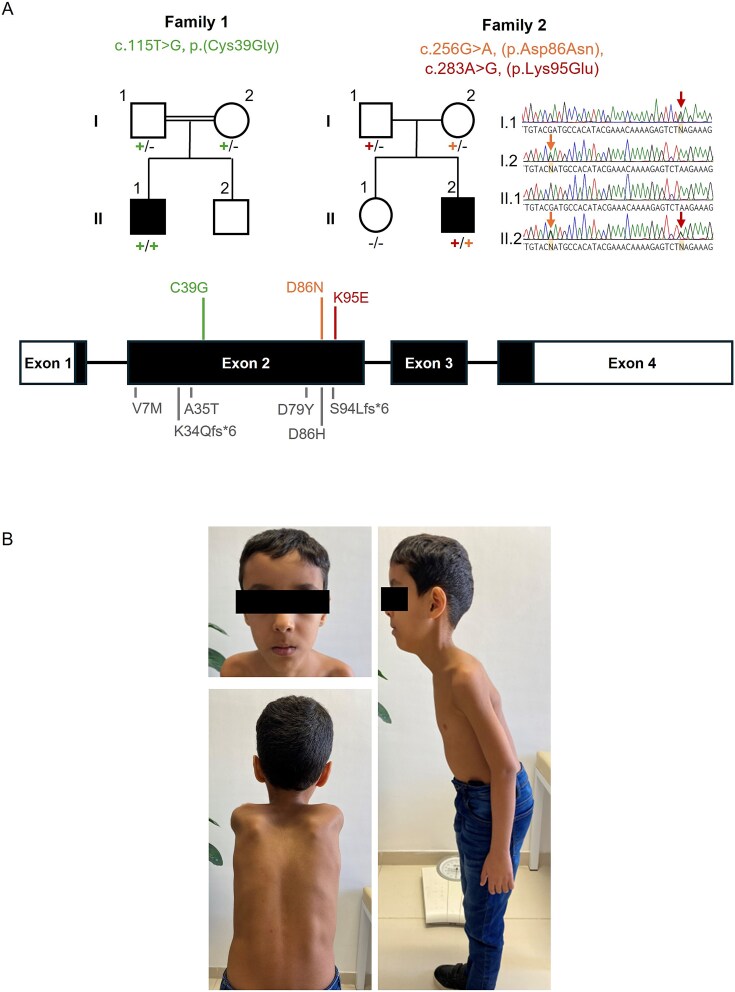
Cofilin-2, encoded by CFL2, is an actin-binding protein essential for regulating actin filament dynamics in skeletal muscle. Biallelic variants in CFL2 are associated with an ultra-rare, early-onset myopathy typically presenting as nemaline myopathy. Only 10 patients have been described to date from five unrelated families. Here, we describe two new cases from two unrelated families. The first proband presented clinically with rigid spine syndrome and a biopsy keeping with nemaline myopathy. The second proband presented with a relatively mild congenital myopathy which became rapidly progressive in the fourth decade, the muscle biopsy showed cytoplasmic bodies, internal nuclei and ringbinden. Exome and genome sequencing revealed three novel biallelic missense variants in CFL2, a homozygous c.115 T > G; p.(Cys39Gly) in the proband of Family 1, and bi-allelic heterozygous c.256G > A:(p.Asp86Asn), and c.283A > G (p.Lys95Glu) variants in the proband of Family 2. We characterised the effects of these substitutions using an in vitro F-actin depolymerisation assay and showed all three were associated with significantly reduced filamentous actin depolymerisation rates compared to the wildtype. Taken together, our findings are highly suggestive of a CFL2-related disease in these patients. Since CFL2-related myopathies are ultrarare, the application of ACMG/AMP guidelines and the diagnostic reportability of CFL2 variants identified in patients remains a challenge. The actin depolymerisation assay may be useful to elucidate the impact and pathogenicity of additional CFL2 variants and has the potential to guide variant classification in future.
期刊介绍:
Human Molecular Genetics concentrates on full-length research papers covering a wide range of topics in all aspects of human molecular genetics. These include:
the molecular basis of human genetic disease
developmental genetics
cancer genetics
neurogenetics
chromosome and genome structure and function
therapy of genetic disease
stem cells in human genetic disease and therapy, including the application of iPS cells
genome-wide association studies
mouse and other models of human diseases
functional genomics
computational genomics
In addition, the journal also publishes research on other model systems for the analysis of genes, especially when there is an obvious relevance to human genetics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


