{"title":"中国桥小脑发育不全家族新型TBC1D23致病变异的鉴定和功能分析。","authors":"Kangyu Liu, Yu Chen, Yunlong Meng, Xinyao Wang, Xingkun Tang, Haining Li, Jianjun Chen, Zilin Zhong","doi":"10.1186/s40246-025-00782-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Pontocerebellar hypoplasia type 11 (PCH11) is an autosomal recessive disorder caused by variants in TBC1D23. The molecular basis for its clinical heterogeneity remains poorly understood. Here, we identified a novel TBC1D23 variant in a Chinese family, investigated its underlying pathogenic mechanisms, and systematically reviewed the clinical phenotypes of all reported cases of PCH11.</p><p><strong>Results: </strong>We identified a novel homozygous frameshift variant, c.511_512delTT (p.F171Qfs*8), in TBC1D23. The patient exhibited a severe phenotype, including marked pontocerebellar hypoplasia, a thinned corpus callosum, global developmental delay, and severe language and motor impairments. Mechanistic studies in a zebrafish model revealed that the mutant transcript partially escaped nonsense-mediated decay (NMD), with expression levels at approximately 50% of the wild-type. In vitro, the resultant truncated protein showed enhanced stability and aberrant cytoplasmic distribution instead of its normal Golgi localization. Furthermore, its expression significantly inhibited cell proliferation.</p><p><strong>Conclusion: </strong>Our study identifies c.511_512delTT as a novel pathogenic variant in TBC1D23. We propose the severe phenotype stems from a primary loss-of-function (LoF), which is likely exacerbated by the cytotoxic effect of the truncated protein produced via partial NMD escape. Our findings suggest this mutant protein exhibits increased stability. This model provides a novel explanation for the phenotypic heterogeneity in PCH11 and expands the mutational spectrum of this disorder.</p>","PeriodicalId":13183,"journal":{"name":"Human Genomics","volume":"19 1","pages":"72"},"PeriodicalIF":4.3000,"publicationDate":"2025-06-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12206363/pdf/","citationCount":"0","resultStr":"{\"title\":\"Identification and functional analysis of a novel TBC1D23 pathogenic variant in a Chinese family with pontocerebellar hypoplasia.\",\"authors\":\"Kangyu Liu, Yu Chen, Yunlong Meng, Xinyao Wang, Xingkun Tang, Haining Li, Jianjun Chen, Zilin Zhong\",\"doi\":\"10.1186/s40246-025-00782-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Pontocerebellar hypoplasia type 11 (PCH11) is an autosomal recessive disorder caused by variants in TBC1D23. The molecular basis for its clinical heterogeneity remains poorly understood. Here, we identified a novel TBC1D23 variant in a Chinese family, investigated its underlying pathogenic mechanisms, and systematically reviewed the clinical phenotypes of all reported cases of PCH11.</p><p><strong>Results: </strong>We identified a novel homozygous frameshift variant, c.511_512delTT (p.F171Qfs*8), in TBC1D23. The patient exhibited a severe phenotype, including marked pontocerebellar hypoplasia, a thinned corpus callosum, global developmental delay, and severe language and motor impairments. Mechanistic studies in a zebrafish model revealed that the mutant transcript partially escaped nonsense-mediated decay (NMD), with expression levels at approximately 50% of the wild-type. In vitro, the resultant truncated protein showed enhanced stability and aberrant cytoplasmic distribution instead of its normal Golgi localization. Furthermore, its expression significantly inhibited cell proliferation.</p><p><strong>Conclusion: </strong>Our study identifies c.511_512delTT as a novel pathogenic variant in TBC1D23. We propose the severe phenotype stems from a primary loss-of-function (LoF), which is likely exacerbated by the cytotoxic effect of the truncated protein produced via partial NMD escape. Our findings suggest this mutant protein exhibits increased stability. This model provides a novel explanation for the phenotypic heterogeneity in PCH11 and expands the mutational spectrum of this disorder.</p>\",\"PeriodicalId\":13183,\"journal\":{\"name\":\"Human Genomics\",\"volume\":\"19 1\",\"pages\":\"72\"},\"PeriodicalIF\":4.3000,\"publicationDate\":\"2025-06-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12206363/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Human Genomics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s40246-025-00782-1\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genomics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s40246-025-00782-1","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Identification and functional analysis of a novel TBC1D23 pathogenic variant in a Chinese family with pontocerebellar hypoplasia.
Background: Pontocerebellar hypoplasia type 11 (PCH11) is an autosomal recessive disorder caused by variants in TBC1D23. The molecular basis for its clinical heterogeneity remains poorly understood. Here, we identified a novel TBC1D23 variant in a Chinese family, investigated its underlying pathogenic mechanisms, and systematically reviewed the clinical phenotypes of all reported cases of PCH11.
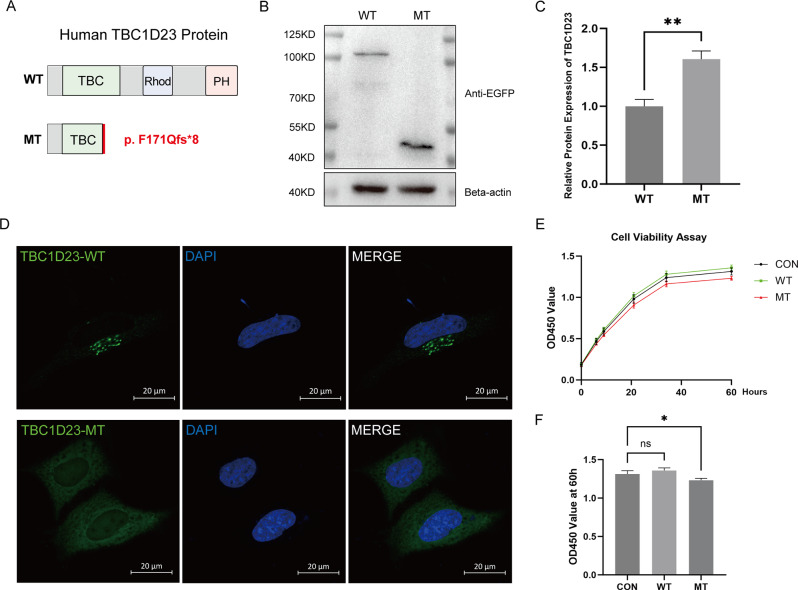
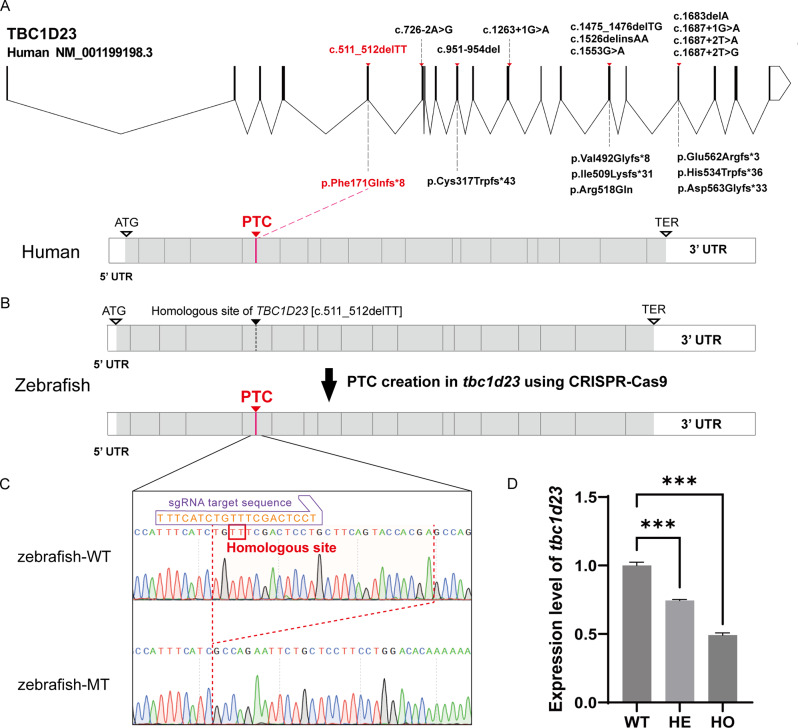
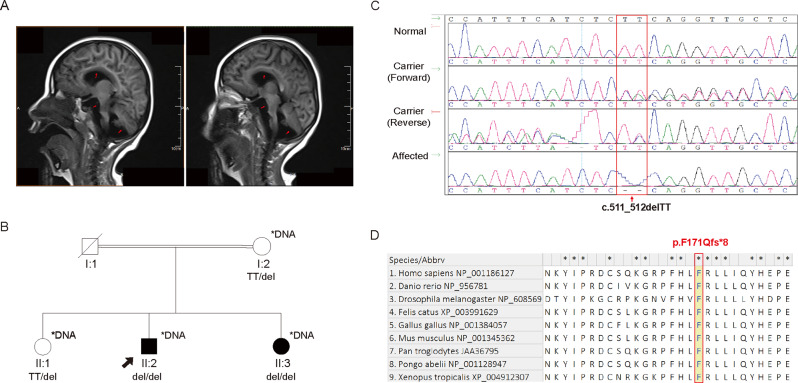
Results: We identified a novel homozygous frameshift variant, c.511_512delTT (p.F171Qfs*8), in TBC1D23. The patient exhibited a severe phenotype, including marked pontocerebellar hypoplasia, a thinned corpus callosum, global developmental delay, and severe language and motor impairments. Mechanistic studies in a zebrafish model revealed that the mutant transcript partially escaped nonsense-mediated decay (NMD), with expression levels at approximately 50% of the wild-type. In vitro, the resultant truncated protein showed enhanced stability and aberrant cytoplasmic distribution instead of its normal Golgi localization. Furthermore, its expression significantly inhibited cell proliferation.
Conclusion: Our study identifies c.511_512delTT as a novel pathogenic variant in TBC1D23. We propose the severe phenotype stems from a primary loss-of-function (LoF), which is likely exacerbated by the cytotoxic effect of the truncated protein produced via partial NMD escape. Our findings suggest this mutant protein exhibits increased stability. This model provides a novel explanation for the phenotypic heterogeneity in PCH11 and expands the mutational spectrum of this disorder.
期刊介绍:
Human Genomics is a peer-reviewed, open access, online journal that focuses on the application of genomic analysis in all aspects of human health and disease, as well as genomic analysis of drug efficacy and safety, and comparative genomics.
Topics covered by the journal include, but are not limited to: pharmacogenomics, genome-wide association studies, genome-wide sequencing, exome sequencing, next-generation deep-sequencing, functional genomics, epigenomics, translational genomics, expression profiling, proteomics, bioinformatics, animal models, statistical genetics, genetic epidemiology, human population genetics and comparative genomics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


