{"title":"利用机器学习模型评估ADMET属性,用于药物发现和开发。","authors":"Magesh Venkataraman, Gopi Chand Rao, Jeevan Karthik Madavareddi, Srinivas Rao Maddi","doi":"10.5599/admet.2772","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and purpose: </strong>The evaluation of ADMET properties remains a critical bottleneck in drug discovery and development, contributing significantly to the high attrition rate of drug candidates. Traditional experimental approaches are often time-consuming, cost-intensive, and limited in scalability. This review aims to investigate how recent advances in machine learning (ML) models are revolutionizing ADMET prediction by enhancing accuracy, reducing experimental burden, and accelerating decision-making during early-stage drug development.</p><p><strong>Experimental approach: </strong>This article systematically examines the current landscape of ML applications in ADMET prediction, including the types of algorithms employed, common molecular descriptors and datasets used, and model development workflows. It also explores public databases, model evaluation metrics, and regulatory considerations relevant to computational toxicology. Emphasis is placed on supervised and deep learning techniques, model validation strategies, and the challenges of data imbalance and model interpretability.</p><p><strong>Key results: </strong>ML-based models have demonstrated significant promise in predicting key ADMET endpoints, outperforming some traditional quantitative structure - activity relationship (QSAR) models. These approaches provide rapid, cost-effective, and reproducible alternatives that integrate seamlessly with existing drug discovery pipelines. Case studies discussed in this review illustrate the successful deployment of ML models for solubility, permeability, metabolism, and toxicity predictions.</p><p><strong>Conclusion: </strong>Machine learning has emerged as a transformative tool in ADMET prediction, offering new opportunities for early risk assessment and compound prioritization. While challenges such as data quality, algorithm transparency, and regulatory acceptance persist, continued integration of ML with experimental pharmacology holds the potential to substantially improve drug development efficiency and reduce late-stage failures.</p>","PeriodicalId":7259,"journal":{"name":"ADMET and DMPK","volume":"13 3","pages":"2772"},"PeriodicalIF":4.3000,"publicationDate":"2025-06-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12205928/pdf/","citationCount":"0","resultStr":"{\"title\":\"Leveraging machine learning models in evaluating ADMET properties for drug discovery and development.\",\"authors\":\"Magesh Venkataraman, Gopi Chand Rao, Jeevan Karthik Madavareddi, Srinivas Rao Maddi\",\"doi\":\"10.5599/admet.2772\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background and purpose: </strong>The evaluation of ADMET properties remains a critical bottleneck in drug discovery and development, contributing significantly to the high attrition rate of drug candidates. Traditional experimental approaches are often time-consuming, cost-intensive, and limited in scalability. This review aims to investigate how recent advances in machine learning (ML) models are revolutionizing ADMET prediction by enhancing accuracy, reducing experimental burden, and accelerating decision-making during early-stage drug development.</p><p><strong>Experimental approach: </strong>This article systematically examines the current landscape of ML applications in ADMET prediction, including the types of algorithms employed, common molecular descriptors and datasets used, and model development workflows. It also explores public databases, model evaluation metrics, and regulatory considerations relevant to computational toxicology. Emphasis is placed on supervised and deep learning techniques, model validation strategies, and the challenges of data imbalance and model interpretability.</p><p><strong>Key results: </strong>ML-based models have demonstrated significant promise in predicting key ADMET endpoints, outperforming some traditional quantitative structure - activity relationship (QSAR) models. These approaches provide rapid, cost-effective, and reproducible alternatives that integrate seamlessly with existing drug discovery pipelines. Case studies discussed in this review illustrate the successful deployment of ML models for solubility, permeability, metabolism, and toxicity predictions.</p><p><strong>Conclusion: </strong>Machine learning has emerged as a transformative tool in ADMET prediction, offering new opportunities for early risk assessment and compound prioritization. While challenges such as data quality, algorithm transparency, and regulatory acceptance persist, continued integration of ML with experimental pharmacology holds the potential to substantially improve drug development efficiency and reduce late-stage failures.</p>\",\"PeriodicalId\":7259,\"journal\":{\"name\":\"ADMET and DMPK\",\"volume\":\"13 3\",\"pages\":\"2772\"},\"PeriodicalIF\":4.3000,\"publicationDate\":\"2025-06-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12205928/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ADMET and DMPK\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.5599/admet.2772\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ADMET and DMPK","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5599/admet.2772","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Leveraging machine learning models in evaluating ADMET properties for drug discovery and development.
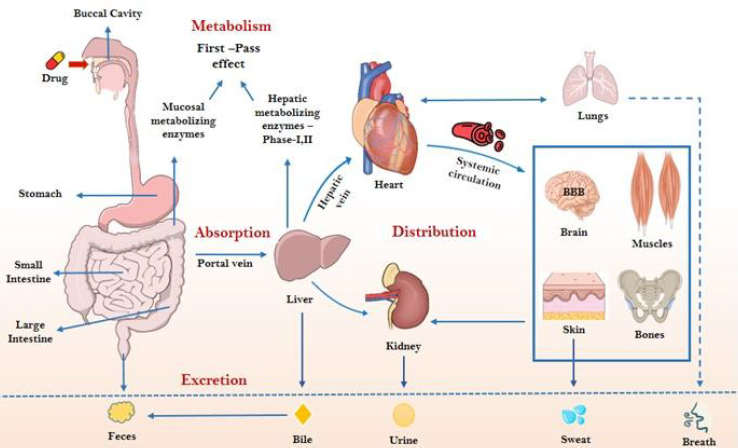
Background and purpose: The evaluation of ADMET properties remains a critical bottleneck in drug discovery and development, contributing significantly to the high attrition rate of drug candidates. Traditional experimental approaches are often time-consuming, cost-intensive, and limited in scalability. This review aims to investigate how recent advances in machine learning (ML) models are revolutionizing ADMET prediction by enhancing accuracy, reducing experimental burden, and accelerating decision-making during early-stage drug development.
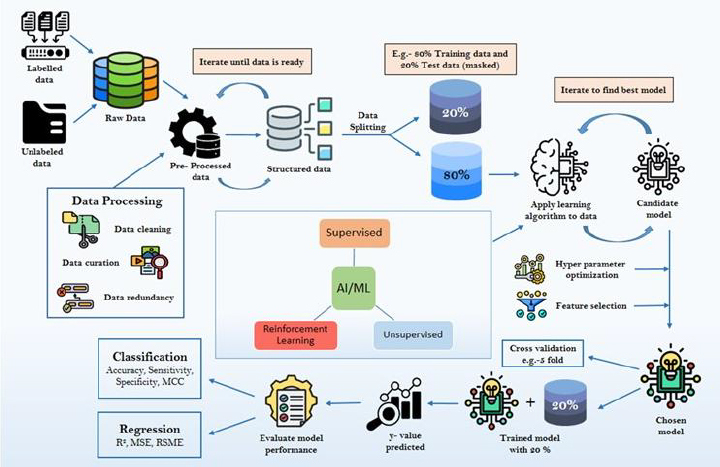
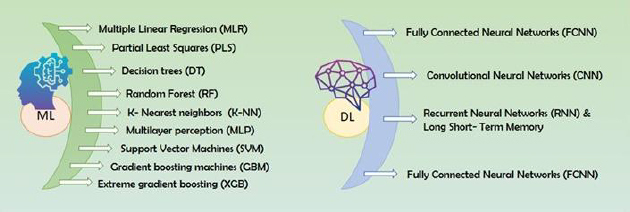
Experimental approach: This article systematically examines the current landscape of ML applications in ADMET prediction, including the types of algorithms employed, common molecular descriptors and datasets used, and model development workflows. It also explores public databases, model evaluation metrics, and regulatory considerations relevant to computational toxicology. Emphasis is placed on supervised and deep learning techniques, model validation strategies, and the challenges of data imbalance and model interpretability.
Key results: ML-based models have demonstrated significant promise in predicting key ADMET endpoints, outperforming some traditional quantitative structure - activity relationship (QSAR) models. These approaches provide rapid, cost-effective, and reproducible alternatives that integrate seamlessly with existing drug discovery pipelines. Case studies discussed in this review illustrate the successful deployment of ML models for solubility, permeability, metabolism, and toxicity predictions.
Conclusion: Machine learning has emerged as a transformative tool in ADMET prediction, offering new opportunities for early risk assessment and compound prioritization. While challenges such as data quality, algorithm transparency, and regulatory acceptance persist, continued integration of ML with experimental pharmacology holds the potential to substantially improve drug development efficiency and reduce late-stage failures.
期刊介绍:
ADMET and DMPK is an open access journal devoted to the rapid dissemination of new and original scientific results in all areas of absorption, distribution, metabolism, excretion, toxicology and pharmacokinetics of drugs. ADMET and DMPK publishes the following types of contributions: - Original research papers - Feature articles - Review articles - Short communications and Notes - Letters to Editors - Book reviews The scope of the Journal involves, but is not limited to, the following areas: - physico-chemical properties of drugs and methods of their determination - drug permeabilities - drug absorption - drug-drug, drug-protein, drug-membrane and drug-DNA interactions - chemical stability and degradations of drugs - instrumental methods in ADMET - drug metablic processes - routes of administration and excretion of drug - pharmacokinetic/pharmacodynamic study - quantitative structure activity/property relationship - ADME/PK modelling - Toxicology screening - Transporter identification and study

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


