{"title":"生化质子转移反应的近似量子化学和机器学习势的基准。","authors":"Guilherme M. Arantes*, and , Jan Řezáč, ","doi":"10.1021/acs.jctc.5c00690","DOIUrl":null,"url":null,"abstract":"<p >Proton transfer reactions are among the most common chemical transformations and are central to enzymatic catalysis and bioenergetic processes. Their mechanisms are often investigated using DFT or approximate quantum chemical methods, whose accuracy directly impacts the reliability of the simulations. Here, a comprehensive set of semiempirical molecular orbital and tight-binding DFT approaches, along with recently developed machine learning (ML) potentials, are benchmarked against high-level MP2 reference data for a curated set of proton transfer reactions representative of biochemical systems. Relative energies, geometries, and dipole moments are evaluated for isolated reactions. Microsolvated reactions are also simulated using a hybrid QM/MM partition. Traditional DFT methods offer high accuracy in general but show markedly larger deviations for proton transfers involving nitrogen-containing groups. Among approximate models, RM1, PM6, PM7, DFTB2-NH, DFTB3, and GFN2-xTB show reasonable accuracy across properties, though their performance varies by chemical group. The ML-corrected (Δ-learning) model PM6-ML improves accuracy for all properties and chemical groups and transfers well to QM/MM simulations. Conversely, standalone ML potentials perform poorly for most reactions. These results provide a basis for evaluating approximate methods and selecting potentials for proton transfer simulations in complex environments.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 14","pages":"7149–7159"},"PeriodicalIF":5.5000,"publicationDate":"2025-06-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12288007/pdf/","citationCount":"0","resultStr":"{\"title\":\"Benchmark of Approximate Quantum Chemical and Machine Learning Potentials for Biochemical Proton Transfer Reactions\",\"authors\":\"Guilherme M. Arantes*, and , Jan Řezáč, \",\"doi\":\"10.1021/acs.jctc.5c00690\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Proton transfer reactions are among the most common chemical transformations and are central to enzymatic catalysis and bioenergetic processes. Their mechanisms are often investigated using DFT or approximate quantum chemical methods, whose accuracy directly impacts the reliability of the simulations. Here, a comprehensive set of semiempirical molecular orbital and tight-binding DFT approaches, along with recently developed machine learning (ML) potentials, are benchmarked against high-level MP2 reference data for a curated set of proton transfer reactions representative of biochemical systems. Relative energies, geometries, and dipole moments are evaluated for isolated reactions. Microsolvated reactions are also simulated using a hybrid QM/MM partition. Traditional DFT methods offer high accuracy in general but show markedly larger deviations for proton transfers involving nitrogen-containing groups. Among approximate models, RM1, PM6, PM7, DFTB2-NH, DFTB3, and GFN2-xTB show reasonable accuracy across properties, though their performance varies by chemical group. The ML-corrected (Δ-learning) model PM6-ML improves accuracy for all properties and chemical groups and transfers well to QM/MM simulations. Conversely, standalone ML potentials perform poorly for most reactions. These results provide a basis for evaluating approximate methods and selecting potentials for proton transfer simulations in complex environments.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"21 14\",\"pages\":\"7149–7159\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-06-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12288007/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00690\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00690","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Benchmark of Approximate Quantum Chemical and Machine Learning Potentials for Biochemical Proton Transfer Reactions
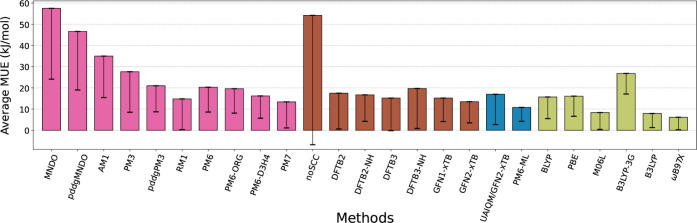
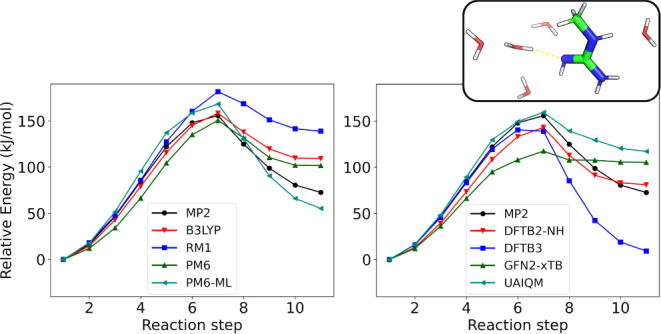
Proton transfer reactions are among the most common chemical transformations and are central to enzymatic catalysis and bioenergetic processes. Their mechanisms are often investigated using DFT or approximate quantum chemical methods, whose accuracy directly impacts the reliability of the simulations. Here, a comprehensive set of semiempirical molecular orbital and tight-binding DFT approaches, along with recently developed machine learning (ML) potentials, are benchmarked against high-level MP2 reference data for a curated set of proton transfer reactions representative of biochemical systems. Relative energies, geometries, and dipole moments are evaluated for isolated reactions. Microsolvated reactions are also simulated using a hybrid QM/MM partition. Traditional DFT methods offer high accuracy in general but show markedly larger deviations for proton transfers involving nitrogen-containing groups. Among approximate models, RM1, PM6, PM7, DFTB2-NH, DFTB3, and GFN2-xTB show reasonable accuracy across properties, though their performance varies by chemical group. The ML-corrected (Δ-learning) model PM6-ML improves accuracy for all properties and chemical groups and transfers well to QM/MM simulations. Conversely, standalone ML potentials perform poorly for most reactions. These results provide a basis for evaluating approximate methods and selecting potentials for proton transfer simulations in complex environments.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


