Nia Pollard, A’Laura C. Hines and Andre Z. Clayborne*,
{"title":"探索量子计算用于金属聚类分析。","authors":"Nia Pollard, A’Laura C. Hines and Andre Z. Clayborne*, ","doi":"10.1021/acs.jpca.5c01404","DOIUrl":null,"url":null,"abstract":"<p >This study explores the application of quantum computing to metal cluster analysis through the development and implementation of a quantum-DFT embedding workflow. Classical computational methods, while transformative, often face limitations in achieving chemical accuracy and computational efficiency, particularly for nanoscale systems. To address these challenges, we integrate the Variational Quantum Eigensolver (VQE) with density functional theory (DFT), leveraging the capabilities of quantum computing aiming to improve the modeling of electronic structures. Aluminum and gold clusters were used as model systems to test the established workflow. The workflow successfully determined electronic properties for aluminum clusters up to Al<sub>7</sub><sup>–</sup>. Although gold clusters were used as a test case to investigate the potential reduction of nitric oxide (NO), memory limitations, the lack of relativistic corrections, and the inability to handle open-shell systems presented challenges that underscore the need for advancements in quantum hardware and algorithms. This proof-of-concept study demonstrates the potential of quantum DFT embedding to advance materials discovery, including applications in catalysis and nanomaterial design, while providing insights into the current limitations of near-term quantum devices.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"129 27","pages":"5923–5930"},"PeriodicalIF":2.8000,"publicationDate":"2025-06-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12257506/pdf/","citationCount":"0","resultStr":"{\"title\":\"Exploring Quantum Computing for Metal Cluster Analysis\",\"authors\":\"Nia Pollard, A’Laura C. Hines and Andre Z. Clayborne*, \",\"doi\":\"10.1021/acs.jpca.5c01404\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >This study explores the application of quantum computing to metal cluster analysis through the development and implementation of a quantum-DFT embedding workflow. Classical computational methods, while transformative, often face limitations in achieving chemical accuracy and computational efficiency, particularly for nanoscale systems. To address these challenges, we integrate the Variational Quantum Eigensolver (VQE) with density functional theory (DFT), leveraging the capabilities of quantum computing aiming to improve the modeling of electronic structures. Aluminum and gold clusters were used as model systems to test the established workflow. The workflow successfully determined electronic properties for aluminum clusters up to Al<sub>7</sub><sup>–</sup>. Although gold clusters were used as a test case to investigate the potential reduction of nitric oxide (NO), memory limitations, the lack of relativistic corrections, and the inability to handle open-shell systems presented challenges that underscore the need for advancements in quantum hardware and algorithms. This proof-of-concept study demonstrates the potential of quantum DFT embedding to advance materials discovery, including applications in catalysis and nanomaterial design, while providing insights into the current limitations of near-term quantum devices.</p>\",\"PeriodicalId\":59,\"journal\":{\"name\":\"The Journal of Physical Chemistry A\",\"volume\":\"129 27\",\"pages\":\"5923–5930\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2025-06-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12257506/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry A\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpca.5c01404\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.5c01404","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Exploring Quantum Computing for Metal Cluster Analysis
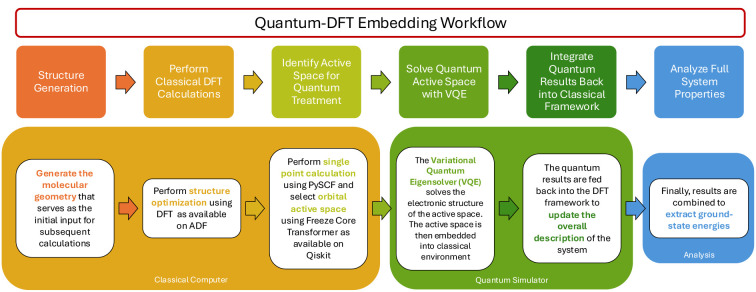
This study explores the application of quantum computing to metal cluster analysis through the development and implementation of a quantum-DFT embedding workflow. Classical computational methods, while transformative, often face limitations in achieving chemical accuracy and computational efficiency, particularly for nanoscale systems. To address these challenges, we integrate the Variational Quantum Eigensolver (VQE) with density functional theory (DFT), leveraging the capabilities of quantum computing aiming to improve the modeling of electronic structures. Aluminum and gold clusters were used as model systems to test the established workflow. The workflow successfully determined electronic properties for aluminum clusters up to Al7–. Although gold clusters were used as a test case to investigate the potential reduction of nitric oxide (NO), memory limitations, the lack of relativistic corrections, and the inability to handle open-shell systems presented challenges that underscore the need for advancements in quantum hardware and algorithms. This proof-of-concept study demonstrates the potential of quantum DFT embedding to advance materials discovery, including applications in catalysis and nanomaterial design, while providing insights into the current limitations of near-term quantum devices.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


