Mary N Chege, Pamela Ferretti, Shasta Webb, Rosaline W Macharia, George Obiero, Joseph Kamau, Susan C Alberts, Jenny Tung, Mercy Y Akinyi, Elizabeth A Archie
{"title":"野生狒狒肠道微生物群中跨季节和社会群体的真核组成。","authors":"Mary N Chege, Pamela Ferretti, Shasta Webb, Rosaline W Macharia, George Obiero, Joseph Kamau, Susan C Alberts, Jenny Tung, Mercy Y Akinyi, Elizabeth A Archie","doi":"10.1186/s42523-025-00436-6","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Animals coexist with complex microbiota, including bacteria, viruses, and eukaryotes (e.g., fungi, protists, and helminths). While high-throughput sequencing is commonly used to characterize bacterial communities in animal microbiota, these methods are less often applied to gut eukaryotic composition. Here we used shotgun metagenomic sequencing to characterize eukaryotic diversity in the microbiomes of wild baboons and tested the degree to which eukaryotic community composition was predicted by host social group membership, sex, age, sequencing depth, and season of sample collection.</p><p><strong>Results: </strong>We analyzed a total of 75 fecal samples collected in 2012 and 2014 from 73 wild baboons in the Amboseli ecosystem in Kenya. DNA from these samples was subjected to shotgun metagenomic sequencing, revealing members of the kingdoms Protista, Chromista, and Fungi in 90.7%, 46.7%, and 20.3% of all samples, respectively (percentages indicate the percent of samples in which each kingdom was observed). Social group membership explained 11.2% of the global diversity in gut eukaryotic species composition, but we did not detect statistically significant effects of season, host age, or host sex. Across samples, the most prevalent protists were Entamoeba coli (74.66% of samples), Enteromonas hominis (53.33% of samples), and Blastocystis subtype 3 (38.66% of samples), while the most prevalent fungi included Pichia manshurica (14.66% of samples), and Ogataea naganishii (6.66% of samples).</p><p><strong>Conclusions: </strong>Protista, Chromista, and Fungi are common members of the gut microbiome of wild baboons. More work on eukaryotic members of primate gut microbiota is important for primate health monitoring and management strategies.</p>","PeriodicalId":72201,"journal":{"name":"Animal microbiome","volume":"7 1","pages":"70"},"PeriodicalIF":4.4000,"publicationDate":"2025-06-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12182654/pdf/","citationCount":"0","resultStr":"{\"title\":\"Eukaryotic composition across seasons and social groups in the gut microbiota of wild baboons.\",\"authors\":\"Mary N Chege, Pamela Ferretti, Shasta Webb, Rosaline W Macharia, George Obiero, Joseph Kamau, Susan C Alberts, Jenny Tung, Mercy Y Akinyi, Elizabeth A Archie\",\"doi\":\"10.1186/s42523-025-00436-6\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Animals coexist with complex microbiota, including bacteria, viruses, and eukaryotes (e.g., fungi, protists, and helminths). While high-throughput sequencing is commonly used to characterize bacterial communities in animal microbiota, these methods are less often applied to gut eukaryotic composition. Here we used shotgun metagenomic sequencing to characterize eukaryotic diversity in the microbiomes of wild baboons and tested the degree to which eukaryotic community composition was predicted by host social group membership, sex, age, sequencing depth, and season of sample collection.</p><p><strong>Results: </strong>We analyzed a total of 75 fecal samples collected in 2012 and 2014 from 73 wild baboons in the Amboseli ecosystem in Kenya. DNA from these samples was subjected to shotgun metagenomic sequencing, revealing members of the kingdoms Protista, Chromista, and Fungi in 90.7%, 46.7%, and 20.3% of all samples, respectively (percentages indicate the percent of samples in which each kingdom was observed). Social group membership explained 11.2% of the global diversity in gut eukaryotic species composition, but we did not detect statistically significant effects of season, host age, or host sex. Across samples, the most prevalent protists were Entamoeba coli (74.66% of samples), Enteromonas hominis (53.33% of samples), and Blastocystis subtype 3 (38.66% of samples), while the most prevalent fungi included Pichia manshurica (14.66% of samples), and Ogataea naganishii (6.66% of samples).</p><p><strong>Conclusions: </strong>Protista, Chromista, and Fungi are common members of the gut microbiome of wild baboons. More work on eukaryotic members of primate gut microbiota is important for primate health monitoring and management strategies.</p>\",\"PeriodicalId\":72201,\"journal\":{\"name\":\"Animal microbiome\",\"volume\":\"7 1\",\"pages\":\"70\"},\"PeriodicalIF\":4.4000,\"publicationDate\":\"2025-06-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12182654/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Animal microbiome\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s42523-025-00436-6\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MICROBIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Animal microbiome","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s42523-025-00436-6","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
Eukaryotic composition across seasons and social groups in the gut microbiota of wild baboons.
Background: Animals coexist with complex microbiota, including bacteria, viruses, and eukaryotes (e.g., fungi, protists, and helminths). While high-throughput sequencing is commonly used to characterize bacterial communities in animal microbiota, these methods are less often applied to gut eukaryotic composition. Here we used shotgun metagenomic sequencing to characterize eukaryotic diversity in the microbiomes of wild baboons and tested the degree to which eukaryotic community composition was predicted by host social group membership, sex, age, sequencing depth, and season of sample collection.
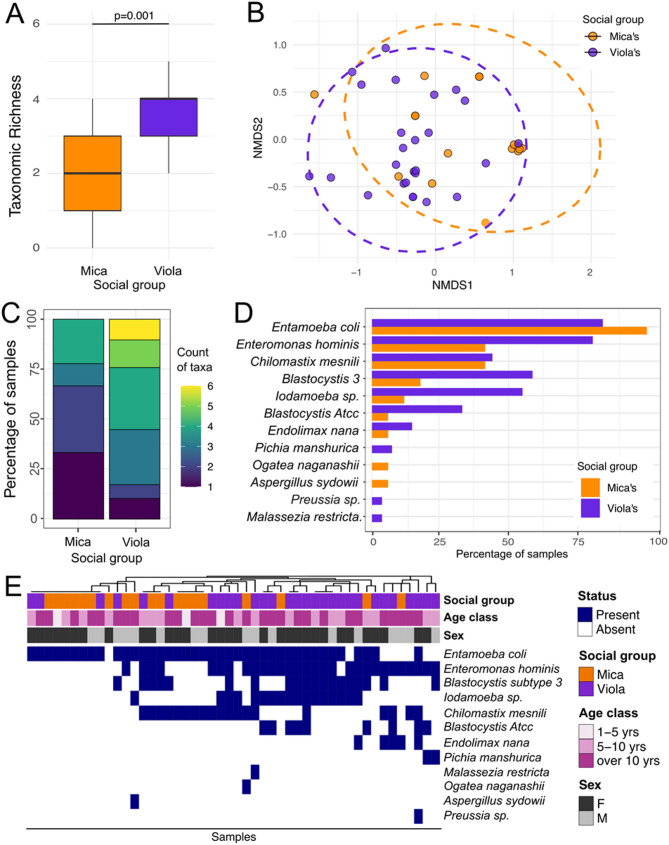
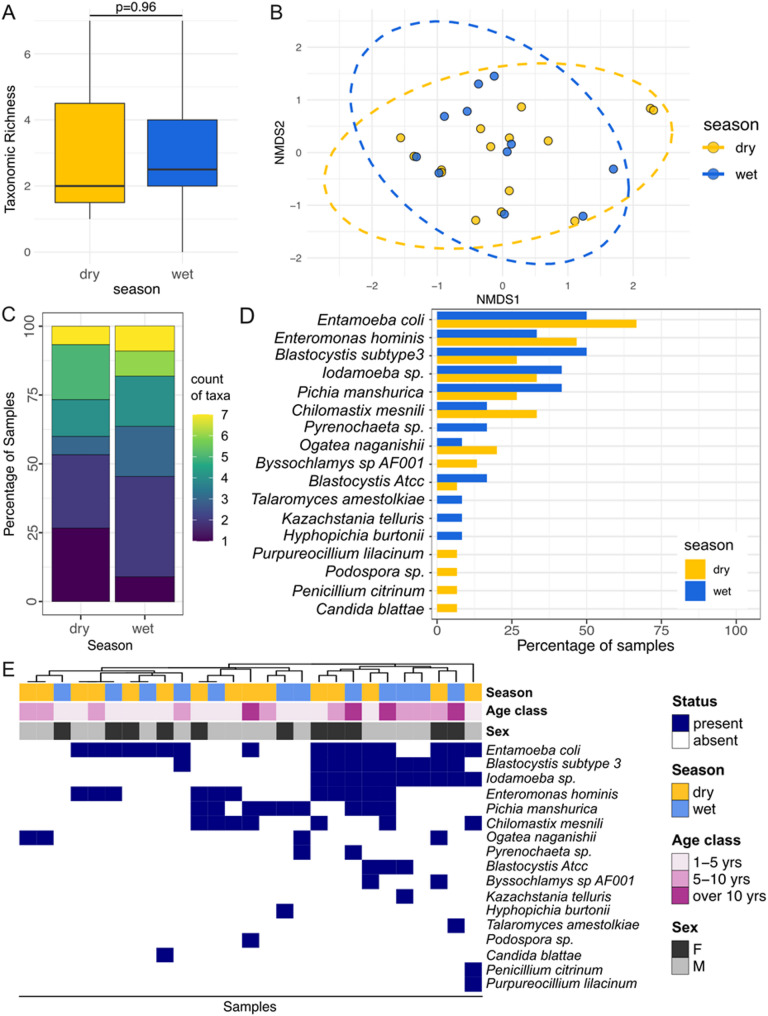
Results: We analyzed a total of 75 fecal samples collected in 2012 and 2014 from 73 wild baboons in the Amboseli ecosystem in Kenya. DNA from these samples was subjected to shotgun metagenomic sequencing, revealing members of the kingdoms Protista, Chromista, and Fungi in 90.7%, 46.7%, and 20.3% of all samples, respectively (percentages indicate the percent of samples in which each kingdom was observed). Social group membership explained 11.2% of the global diversity in gut eukaryotic species composition, but we did not detect statistically significant effects of season, host age, or host sex. Across samples, the most prevalent protists were Entamoeba coli (74.66% of samples), Enteromonas hominis (53.33% of samples), and Blastocystis subtype 3 (38.66% of samples), while the most prevalent fungi included Pichia manshurica (14.66% of samples), and Ogataea naganishii (6.66% of samples).
Conclusions: Protista, Chromista, and Fungi are common members of the gut microbiome of wild baboons. More work on eukaryotic members of primate gut microbiota is important for primate health monitoring and management strategies.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


