Yuanxin Cao, Henrik P. H. Wong, Yi Zhang, Jim Warwicker, Sam Hay and Sam P. de Visser*,
{"title":"亚甲基插入C-N键:非血红素铁加氧酶生物合成脱氢磷霉素的机制","authors":"Yuanxin Cao, Henrik P. H. Wong, Yi Zhang, Jim Warwicker, Sam Hay and Sam P. de Visser*, ","doi":"10.1021/acscatal.5c0326810.1021/acscatal.5c03268","DOIUrl":null,"url":null,"abstract":"<p >The nonheme iron dioxygenase dehydrofosmidomycin synthase D (DfmD) performs an unusual catalytic reaction mechanism with an initial desaturation followed by <i>N</i>-demethylation and the insertion of a CH<sub>2</sub> group into a C–N bond. However, the reaction process is challenging synthetically and an enzymatic approach by a single protein may have applications in biotechnology for the biosynthesis of drug molecules. Little is known regarding the details of the reaction mechanism and several possible mechanisms have been proposed. To resolve the controversy, we performed a detailed computational study on the desaturation and rearrangement steps of 2-(trimethylammonio)ethylphosphonate by DfmD using two molecules of O<sub>2</sub> and α-ketoglutarate to form methyldehydrofosmidomycin. Interestingly, 2-(trimethylammonio)ethylphosphonate dioxygenase (TmpA) activates the same substrate but produces C<sub>1</sub>-hydroxylation products instead and only runs one catalytic cycle with the substrate. Our computational study uses molecular dynamics and quantum mechanics calculations and focuses on the comparison of the structure, mechanism, and function of the two enzymes. The initial molecular dynamics simulation shows that the substrate is tightly bound to the substrate-binding pocket although its position differs from that in TmpA. We created enzymatic models and studied the substrate-binding and reaction mechanisms leading to products and potential byproducts for the two successive catalytic cycles with an iron(IV)-oxo species. Molecular dynamics simulations confirm that the product after the first cycle does not escape the protein in DfmD. Our calculations confirm experimental product distributions and show a dominant C<sub>1</sub>-hydroxylation mechanism for TmpA, whereas DfmD reacts through a dominant desaturation of the C<sub>1</sub>–C<sub>2</sub> bond with two consecutive hydrogen atom abstraction steps from the C<sub>1</sub>–H and C<sub>2</sub>–H bonds. A second oxidation cycle with O<sub>2</sub> and α-ketoglutarate starts with hydrogen atom abstraction from the <i>N</i>-methyl group followed by CH<sub>2</sub> insertion into the C–N bond and OH rebound to form methyldehydrofosmidomycin products. We analyzed the optimized geometries, charge distributions, and substrate mobility patterns and concluded that the active site of TmpA binds the substrate strongly through salt bridges and hydrogen bonding interactions that guide the selectivity to C<sub>1</sub>-hydroxylation, while the different polarity and local electric field directions of the binding pocket in DfmD guide the reaction to desaturation instead.</p>","PeriodicalId":9,"journal":{"name":"ACS Catalysis ","volume":"15 12","pages":"10828–10846 10828–10846"},"PeriodicalIF":13.1000,"publicationDate":"2025-06-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Methylene Group Insertion into a C–N Bond: The Mechanism for the Biosynthesis of Dehydrofosmidomycin by a Nonheme Iron Oxygenase\",\"authors\":\"Yuanxin Cao, Henrik P. H. Wong, Yi Zhang, Jim Warwicker, Sam Hay and Sam P. de Visser*, \",\"doi\":\"10.1021/acscatal.5c0326810.1021/acscatal.5c03268\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The nonheme iron dioxygenase dehydrofosmidomycin synthase D (DfmD) performs an unusual catalytic reaction mechanism with an initial desaturation followed by <i>N</i>-demethylation and the insertion of a CH<sub>2</sub> group into a C–N bond. However, the reaction process is challenging synthetically and an enzymatic approach by a single protein may have applications in biotechnology for the biosynthesis of drug molecules. Little is known regarding the details of the reaction mechanism and several possible mechanisms have been proposed. To resolve the controversy, we performed a detailed computational study on the desaturation and rearrangement steps of 2-(trimethylammonio)ethylphosphonate by DfmD using two molecules of O<sub>2</sub> and α-ketoglutarate to form methyldehydrofosmidomycin. Interestingly, 2-(trimethylammonio)ethylphosphonate dioxygenase (TmpA) activates the same substrate but produces C<sub>1</sub>-hydroxylation products instead and only runs one catalytic cycle with the substrate. Our computational study uses molecular dynamics and quantum mechanics calculations and focuses on the comparison of the structure, mechanism, and function of the two enzymes. The initial molecular dynamics simulation shows that the substrate is tightly bound to the substrate-binding pocket although its position differs from that in TmpA. We created enzymatic models and studied the substrate-binding and reaction mechanisms leading to products and potential byproducts for the two successive catalytic cycles with an iron(IV)-oxo species. Molecular dynamics simulations confirm that the product after the first cycle does not escape the protein in DfmD. Our calculations confirm experimental product distributions and show a dominant C<sub>1</sub>-hydroxylation mechanism for TmpA, whereas DfmD reacts through a dominant desaturation of the C<sub>1</sub>–C<sub>2</sub> bond with two consecutive hydrogen atom abstraction steps from the C<sub>1</sub>–H and C<sub>2</sub>–H bonds. A second oxidation cycle with O<sub>2</sub> and α-ketoglutarate starts with hydrogen atom abstraction from the <i>N</i>-methyl group followed by CH<sub>2</sub> insertion into the C–N bond and OH rebound to form methyldehydrofosmidomycin products. We analyzed the optimized geometries, charge distributions, and substrate mobility patterns and concluded that the active site of TmpA binds the substrate strongly through salt bridges and hydrogen bonding interactions that guide the selectivity to C<sub>1</sub>-hydroxylation, while the different polarity and local electric field directions of the binding pocket in DfmD guide the reaction to desaturation instead.</p>\",\"PeriodicalId\":9,\"journal\":{\"name\":\"ACS Catalysis \",\"volume\":\"15 12\",\"pages\":\"10828–10846 10828–10846\"},\"PeriodicalIF\":13.1000,\"publicationDate\":\"2025-06-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ACS Catalysis \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acscatal.5c03268\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Catalysis ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acscatal.5c03268","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Methylene Group Insertion into a C–N Bond: The Mechanism for the Biosynthesis of Dehydrofosmidomycin by a Nonheme Iron Oxygenase
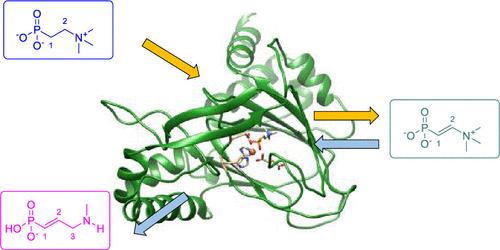
The nonheme iron dioxygenase dehydrofosmidomycin synthase D (DfmD) performs an unusual catalytic reaction mechanism with an initial desaturation followed by N-demethylation and the insertion of a CH2 group into a C–N bond. However, the reaction process is challenging synthetically and an enzymatic approach by a single protein may have applications in biotechnology for the biosynthesis of drug molecules. Little is known regarding the details of the reaction mechanism and several possible mechanisms have been proposed. To resolve the controversy, we performed a detailed computational study on the desaturation and rearrangement steps of 2-(trimethylammonio)ethylphosphonate by DfmD using two molecules of O2 and α-ketoglutarate to form methyldehydrofosmidomycin. Interestingly, 2-(trimethylammonio)ethylphosphonate dioxygenase (TmpA) activates the same substrate but produces C1-hydroxylation products instead and only runs one catalytic cycle with the substrate. Our computational study uses molecular dynamics and quantum mechanics calculations and focuses on the comparison of the structure, mechanism, and function of the two enzymes. The initial molecular dynamics simulation shows that the substrate is tightly bound to the substrate-binding pocket although its position differs from that in TmpA. We created enzymatic models and studied the substrate-binding and reaction mechanisms leading to products and potential byproducts for the two successive catalytic cycles with an iron(IV)-oxo species. Molecular dynamics simulations confirm that the product after the first cycle does not escape the protein in DfmD. Our calculations confirm experimental product distributions and show a dominant C1-hydroxylation mechanism for TmpA, whereas DfmD reacts through a dominant desaturation of the C1–C2 bond with two consecutive hydrogen atom abstraction steps from the C1–H and C2–H bonds. A second oxidation cycle with O2 and α-ketoglutarate starts with hydrogen atom abstraction from the N-methyl group followed by CH2 insertion into the C–N bond and OH rebound to form methyldehydrofosmidomycin products. We analyzed the optimized geometries, charge distributions, and substrate mobility patterns and concluded that the active site of TmpA binds the substrate strongly through salt bridges and hydrogen bonding interactions that guide the selectivity to C1-hydroxylation, while the different polarity and local electric field directions of the binding pocket in DfmD guide the reaction to desaturation instead.
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


