Nahyun Chi, Jungim Han, Joonghee Won and Jun Soo Kim*,
{"title":"直接分子动力学模拟有机晶体熔点预测","authors":"Nahyun Chi, Jungim Han, Joonghee Won and Jun Soo Kim*, ","doi":"10.1021/acs.cgd.4c0175310.1021/acs.cgd.4c01753","DOIUrl":null,"url":null,"abstract":"<p >Accurate melting point prediction is essential for investigating the molecular mechanisms of crystal growth and melting using molecular dynamics (MD) simulations. Here, we assess melting point predictions from direct MD simulations of nitromethane and acetic acid. This study has three objectives: to evaluate popular force fields (CGenFF, OPLS, GAFF), to assess various MD approaches (simulations of solid/liquid, vapor/solid/liquid/vapor, vapor/solid/vapor, and solid alone), and to compare the crystal growth and melting of both compounds, focusing specifically on the time scale and anisotropy. Our results indicate that none of the popular force fields accurately predict melting points, highlighting the need for improvement. All MD simulation approaches yielded consistent melting points of either compound, except for the solid-alone simulation, while continuous heating of the vapor/solid/vapor system proved effective. The time scales of crystal growth and melting differ significantly between the molecules: 20 ns for nitromethane and 200 ns for acetic acid. Anisotropy in crystal growth and melting is non-negligible and much more pronounced for acetic acid compared to nitromethane. These findings offer practical considerations for simulating melting phenomena in molecular crystals using MD.</p>","PeriodicalId":34,"journal":{"name":"Crystal Growth & Design","volume":"25 12","pages":"4169–4177 4169–4177"},"PeriodicalIF":3.4000,"publicationDate":"2025-06-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Melting Point Prediction of Organic Crystals Using Direct Molecular Dynamics Simulations\",\"authors\":\"Nahyun Chi, Jungim Han, Joonghee Won and Jun Soo Kim*, \",\"doi\":\"10.1021/acs.cgd.4c0175310.1021/acs.cgd.4c01753\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Accurate melting point prediction is essential for investigating the molecular mechanisms of crystal growth and melting using molecular dynamics (MD) simulations. Here, we assess melting point predictions from direct MD simulations of nitromethane and acetic acid. This study has three objectives: to evaluate popular force fields (CGenFF, OPLS, GAFF), to assess various MD approaches (simulations of solid/liquid, vapor/solid/liquid/vapor, vapor/solid/vapor, and solid alone), and to compare the crystal growth and melting of both compounds, focusing specifically on the time scale and anisotropy. Our results indicate that none of the popular force fields accurately predict melting points, highlighting the need for improvement. All MD simulation approaches yielded consistent melting points of either compound, except for the solid-alone simulation, while continuous heating of the vapor/solid/vapor system proved effective. The time scales of crystal growth and melting differ significantly between the molecules: 20 ns for nitromethane and 200 ns for acetic acid. Anisotropy in crystal growth and melting is non-negligible and much more pronounced for acetic acid compared to nitromethane. These findings offer practical considerations for simulating melting phenomena in molecular crystals using MD.</p>\",\"PeriodicalId\":34,\"journal\":{\"name\":\"Crystal Growth & Design\",\"volume\":\"25 12\",\"pages\":\"4169–4177 4169–4177\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2025-06-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Crystal Growth & Design\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.cgd.4c01753\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Crystal Growth & Design","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.cgd.4c01753","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Melting Point Prediction of Organic Crystals Using Direct Molecular Dynamics Simulations
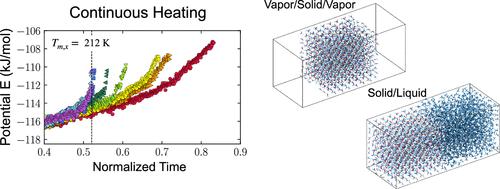
Accurate melting point prediction is essential for investigating the molecular mechanisms of crystal growth and melting using molecular dynamics (MD) simulations. Here, we assess melting point predictions from direct MD simulations of nitromethane and acetic acid. This study has three objectives: to evaluate popular force fields (CGenFF, OPLS, GAFF), to assess various MD approaches (simulations of solid/liquid, vapor/solid/liquid/vapor, vapor/solid/vapor, and solid alone), and to compare the crystal growth and melting of both compounds, focusing specifically on the time scale and anisotropy. Our results indicate that none of the popular force fields accurately predict melting points, highlighting the need for improvement. All MD simulation approaches yielded consistent melting points of either compound, except for the solid-alone simulation, while continuous heating of the vapor/solid/vapor system proved effective. The time scales of crystal growth and melting differ significantly between the molecules: 20 ns for nitromethane and 200 ns for acetic acid. Anisotropy in crystal growth and melting is non-negligible and much more pronounced for acetic acid compared to nitromethane. These findings offer practical considerations for simulating melting phenomena in molecular crystals using MD.
期刊介绍:
The aim of Crystal Growth & Design is to stimulate crossfertilization of knowledge among scientists and engineers working in the fields of crystal growth, crystal engineering, and the industrial application of crystalline materials.
Crystal Growth & Design publishes theoretical and experimental studies of the physical, chemical, and biological phenomena and processes related to the design, growth, and application of crystalline materials. Synergistic approaches originating from different disciplines and technologies and integrating the fields of crystal growth, crystal engineering, intermolecular interactions, and industrial application are encouraged.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


