基于脂肪酸结合蛋白I型(FABP I型)基因和线粒体DNA系统发育分析的肝片形吸虫与 rkiye虫的准确种分
IF 2.6
4区 医学
Q3 INFECTIOUS DISEASES
引用次数: 0
摘要
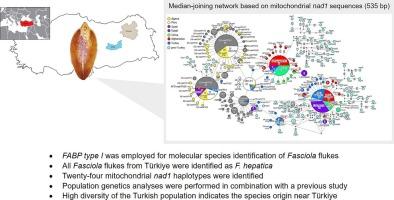
基于强大的核蛋白编码基因标记的准确物种区分对于片形吸虫至关重要,因为在亚洲存在肝吸虫、巨型吸虫和源自两种种间杂交的杂交片形吸虫。采用靶向磷酸烯醇丙酮酸羧激酶(pepck)的多重聚合酶链反应(PCR)和靶向DNA聚合酶delta (pold)的PCR-限制性片段长度多态性(RFLP)技术对片形吸虫进行分子鉴定。然而,这些方法已经证明了局限性,包括错误识别。在这项研究中,我们旨在探讨多重PCR使用脂肪酸结合蛋白I型(FABP I型)基因作为物种鉴定标记的可靠性。在本研究中,使用FABPⅰ型对75个肝吸虫进行了鉴定。FABPⅰ型对肝吸虫的种类鉴定比以往报道的标记物pepck和pold更为准确和有用,因为没有发现鉴别错误,而pepck和pold分别对5个和13个吸虫出现了误诊和扩增失败。我们还试图根据线粒体DNA NADH脱氢酶亚基1 (nad1)基因的核苷酸序列分析土耳其吸片吸虫的系统发育。值得注意的是,土耳其肝f菌种群比先前研究的参考种群显示出更大的遗传多样性,这表明土耳其种群的年龄要大得多。这支持了先前提出的肝原胞菌起源于毗邻本文章由计算机程序翻译,如有差异,请以英文原文为准。
Accurate species discrimination of Fasciola hepatica from Türkiye based on the fatty acid binding protein type I (FABP type I) gene and phylogenetic analysis using mitochondrial DNA
Accurate species discrimination based on a robust nuclear protein-coding gene marker is essential for Fasciola spp. because of the presence of F. hepatica, F. gigantica, and hybrid Fasciola flukes that originate from the interspecific hybridization between the two species in Asia. Molecular identification of Fasciola species uses multiplex polymerase chain reaction (PCR) targeting phosphoenolpyruvate carboxykinase (pepck) and PCR-restriction fragment length polymorphism (RFLP) targeting DNA polymerase delta (pold). However, these methods have demonstrated limitations, including misidentification errors. In this study, we aimed to investigate the reliability of multiplex PCR using the fatty acid binding protein type I (FABP type I) gene as a species identification marker. In this study, 75 liver flukes were determined as F. hepatica using FABP type I. FABP type I was more accurate and useful for species identification of F. hepatica than previously reported markers, pepck and pold, as no discrimination errors were observed, whereas misdiagnosis and amplification failure occurred in pepck and pold for five and 13 flukes, respectively. We also aimed to analyze the phylogeny of Turkish Fasciola flukes based on nucleotide sequences of the NADH dehydrogenase subunit 1 (nad1) gene of mitochondrial DNA. Notably, the Turkish F. hepatica population showed greater genetic diversity than the reference populations from the previous studies, suggesting that the Turkish population is much older. This supports the idea that F. hepatica originated in the Fertile Crescent, the area adjacent to Türkiye, as proposed previously.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Infection Genetics and Evolution
医学-传染病学
CiteScore
8.40
自引率
0.00%
发文量
215
审稿时长
82 days
期刊介绍:
(aka Journal of Molecular Epidemiology and Evolutionary Genetics of Infectious Diseases -- MEEGID)
Infectious diseases constitute one of the main challenges to medical science in the coming century. The impressive development of molecular megatechnologies and of bioinformatics have greatly increased our knowledge of the evolution, transmission and pathogenicity of infectious diseases. Research has shown that host susceptibility to many infectious diseases has a genetic basis. Furthermore, much is now known on the molecular epidemiology, evolution and virulence of pathogenic agents, as well as their resistance to drugs, vaccines, and antibiotics. Equally, research on the genetics of disease vectors has greatly improved our understanding of their systematics, has increased our capacity to identify target populations for control or intervention, and has provided detailed information on the mechanisms of insecticide resistance.
However, the genetics and evolutionary biology of hosts, pathogens and vectors have tended to develop as three separate fields of research. This artificial compartmentalisation is of concern due to our growing appreciation of the strong co-evolutionary interactions among hosts, pathogens and vectors.
Infection, Genetics and Evolution and its companion congress [MEEGID](http://www.meegidconference.com/) (for Molecular Epidemiology and Evolutionary Genetics of Infectious Diseases) are the main forum acting for the cross-fertilization between evolutionary science and biomedical research on infectious diseases.
Infection, Genetics and Evolution is the only journal that welcomes articles dealing with the genetics and evolutionary biology of hosts, pathogens and vectors, and coevolution processes among them in relation to infection and disease manifestation. All infectious models enter the scope of the journal, including pathogens of humans, animals and plants, either parasites, fungi, bacteria, viruses or prions. The journal welcomes articles dealing with genetics, population genetics, genomics, postgenomics, gene expression, evolutionary biology, population dynamics, mathematical modeling and bioinformatics. We also provide many author benefits, such as free PDFs, a liberal copyright policy, special discounts on Elsevier publications and much more. Please click here for more information on our author services .
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


