一个中国家庭中由致病性METTL5剪接突变引起的小头症相关的整体发育迟缓
IF 2.5
3区 生物学
Q2 GENETICS & HEREDITY
引用次数: 0
摘要
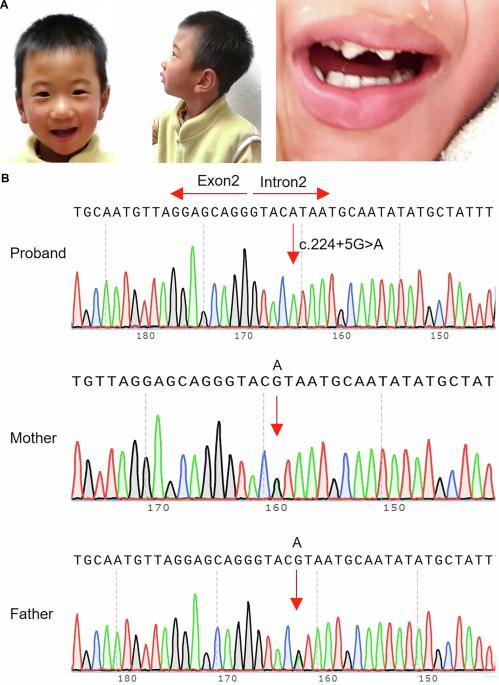
小头症相关的全面发育迟缓(GDD)和智力残疾(ID)以广泛的神经发育障碍为特征,包括多种病因。METTL5是参与18S rRNA甲基化的关键成分,由于其在小头畸形相关的GDD和ID发病机制中的关键作用而引起了相当大的关注。我们对一名2岁女童进行了全面的体格检查和发育评估,该女童表现为GDD和原发性小头畸形。采用全外显子组测序(WES)鉴定致病变异,Sanger测序证实了该突变。为了进一步研究该突变的致病性,采用了短基因剪接实验、体内RT-PCR和生物信息学分析。WES在先证者中鉴定出一个METTL5纯合内含子突变(NM_014168.4: c.224+5 G > a)。Sanger测序进一步证实了该家族的突变。miniigene分析和体内RT-PCR分析显示外显子2跳变,导致突变序列中115 bp的缺失。生物信息学分析证实了该突变的致病性。本研究首次报道了METTL5基因纯合突变(c.224+5 G > a)导致一个中国家庭小头症相关GDD。同时,该报告证实了内含子突变的致病性,扩大了METTL5基因的突变谱。因此,本研究有助于我们了解METTL5在GDD中的作用,为GDD的预防提供理论基础。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Microcephaly-related global developmental delay caused by a pathogenic METTL5 splicing mutation in a Chinese family
Microcephaly-related global developmental delay (GDD) and intellectual disability (ID) are characterized by a broad spectrum of neurodevelopmental impairments and encompass a multitude of causal factors. METTL5, a critical component involved in 18S rRNA methylation, has garnered considerable attention owing to its pivotal role in the pathogenesis of GDD and ID associated with microcephaly. A comprehensive physical examination and developmental assessment were performed for a 2-year-old girl presenting with symptoms of GDD and primary microcephaly. Whole-exome sequencing (WES) was performed to identify the pathogenic variant, and Sanger sequencing confirmed the mutation. To further investigate the pathogenicity of the mutation, minigene splicing assays, in vivo RT-PCR and bioinformatics analysis were employed. The WES identified a METTL5 homozygous intron mutation (NM_014168.4: c.224+5 G > A) in the proband. Sanger sequencing further validated the mutation in the family. Minigene assays and in vivo RT-PCR assays demonstrated exon 2 skipping, resulting in a 115-bp deletion in the mutated sequence. Bioinformatics analysis confirmed the pathogenicity of the mutation. For the first time, this study reported that a homozygous mutation (c.224+5 G > A) in the METTL5 gene led to microcephaly-related GDD in a Chinese family. Meantime, the report has validated the pathogenicity of intronic mutations and expanded the mutational spectrum of the METTL5 gene. Thus, this study aids our understanding of the role of METTL5 in GDD and provides a theoretical foundation for the prevention of this disease.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Journal of Human Genetics
生物-遗传学
CiteScore
7.20
自引率
0.00%
发文量
101
审稿时长
4-8 weeks
期刊介绍:
The Journal of Human Genetics is an international journal publishing articles on human genetics, including medical genetics and human genome analysis. It covers all aspects of human genetics, including molecular genetics, clinical genetics, behavioral genetics, immunogenetics, pharmacogenomics, population genetics, functional genomics, epigenetics, genetic counseling and gene therapy.
Articles on the following areas are especially welcome: genetic factors of monogenic and complex disorders, genome-wide association studies, genetic epidemiology, cancer genetics, personal genomics, genotype-phenotype relationships and genome diversity.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


