{"title":"OpenOrbitalOptimizer─一个用于自一致字段计算的可重用开源库。","authors":"Susi Lehtola*, and , Lori A. Burns, ","doi":"10.1021/acs.jpca.5c02110","DOIUrl":null,"url":null,"abstract":"<p >According to the modern paradigms of software engineering, standard tasks are best accomplished by reusable open source libraries. We describe OpenOrbitalOptimizer: a reusable open source C++ library for the iterative solution of coupled self-consistent field (SCF) equations <b><i>F</i></b><sub><i>p</i></sub><sup>σ</sup>({<b><i>C</i></b><sub><i>p</i></sub><sup>σ</sup>}) <b><i>C</i></b><sub><i>p</i></sub><sup>σ</sup> = <b><i>C</i></b><sub><i>p</i></sub><sup>σ</sup><b><i>E</i></b><sub><i>p</i></sub><sup>σ</sup> for an arbitrary number of particle types <i>p</i> and symmetries. Although OpenOrbitalOptimizer is a new project, it already implements standard algorithms for solving SCF equations: Pulay’s direct inversion in the iterative subspace (DIIS), energy DIIS (EDIIS), augmented DIIS (ADIIS), and the optimal damping algorithm (ODA). The library was designed as an easy way to introduce state-of-the-art convergence accelerators in a number of legacy programs. It is easy to interface with various programs, as it only requires a function to evaluate the total energy <i>E</i> and Fock matrices {<b><i>F</i></b><sub><i>p</i></sub><sup>σ</sup>} for a given set of orbitals {<b><i>C</i></b><sub><i>p</i></sub><sup>σ</sup>}. The only assumption behind the library is that one is able to easily store Fock and orbital matrices in memory, and to diagonalize the Fock matrices in full, which is the case in the overwhelming majority of quantum chemistry applications. We exemplify the library with nuclear-electronic orbital (NEO) calculations of protonated water clusters with Gaussian-type orbital basis sets. We find that a minimal-basis protonic guess works well, and that the stepwise SCF algorithm requires less computational time than the simultaneous SCF algorithm.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"129 25","pages":"5651–5664"},"PeriodicalIF":2.8000,"publicationDate":"2025-06-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12207584/pdf/","citationCount":"0","resultStr":"{\"title\":\"OpenOrbitalOptimizer─A Reusable Open Source Library for Self-Consistent Field Calculations\",\"authors\":\"Susi Lehtola*, and , Lori A. Burns, \",\"doi\":\"10.1021/acs.jpca.5c02110\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >According to the modern paradigms of software engineering, standard tasks are best accomplished by reusable open source libraries. We describe OpenOrbitalOptimizer: a reusable open source C++ library for the iterative solution of coupled self-consistent field (SCF) equations <b><i>F</i></b><sub><i>p</i></sub><sup>σ</sup>({<b><i>C</i></b><sub><i>p</i></sub><sup>σ</sup>}) <b><i>C</i></b><sub><i>p</i></sub><sup>σ</sup> = <b><i>C</i></b><sub><i>p</i></sub><sup>σ</sup><b><i>E</i></b><sub><i>p</i></sub><sup>σ</sup> for an arbitrary number of particle types <i>p</i> and symmetries. Although OpenOrbitalOptimizer is a new project, it already implements standard algorithms for solving SCF equations: Pulay’s direct inversion in the iterative subspace (DIIS), energy DIIS (EDIIS), augmented DIIS (ADIIS), and the optimal damping algorithm (ODA). The library was designed as an easy way to introduce state-of-the-art convergence accelerators in a number of legacy programs. It is easy to interface with various programs, as it only requires a function to evaluate the total energy <i>E</i> and Fock matrices {<b><i>F</i></b><sub><i>p</i></sub><sup>σ</sup>} for a given set of orbitals {<b><i>C</i></b><sub><i>p</i></sub><sup>σ</sup>}. The only assumption behind the library is that one is able to easily store Fock and orbital matrices in memory, and to diagonalize the Fock matrices in full, which is the case in the overwhelming majority of quantum chemistry applications. We exemplify the library with nuclear-electronic orbital (NEO) calculations of protonated water clusters with Gaussian-type orbital basis sets. We find that a minimal-basis protonic guess works well, and that the stepwise SCF algorithm requires less computational time than the simultaneous SCF algorithm.</p>\",\"PeriodicalId\":59,\"journal\":{\"name\":\"The Journal of Physical Chemistry A\",\"volume\":\"129 25\",\"pages\":\"5651–5664\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2025-06-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12207584/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry A\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpca.5c02110\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.5c02110","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
OpenOrbitalOptimizer─A Reusable Open Source Library for Self-Consistent Field Calculations
According to the modern paradigms of software engineering, standard tasks are best accomplished by reusable open source libraries. We describe OpenOrbitalOptimizer: a reusable open source C++ library for the iterative solution of coupled self-consistent field (SCF) equations Fpσ({Cpσ}) Cpσ = CpσEpσ for an arbitrary number of particle types p and symmetries. Although OpenOrbitalOptimizer is a new project, it already implements standard algorithms for solving SCF equations: Pulay’s direct inversion in the iterative subspace (DIIS), energy DIIS (EDIIS), augmented DIIS (ADIIS), and the optimal damping algorithm (ODA). The library was designed as an easy way to introduce state-of-the-art convergence accelerators in a number of legacy programs. It is easy to interface with various programs, as it only requires a function to evaluate the total energy E and Fock matrices {Fpσ} for a given set of orbitals {Cpσ}. The only assumption behind the library is that one is able to easily store Fock and orbital matrices in memory, and to diagonalize the Fock matrices in full, which is the case in the overwhelming majority of quantum chemistry applications. We exemplify the library with nuclear-electronic orbital (NEO) calculations of protonated water clusters with Gaussian-type orbital basis sets. We find that a minimal-basis protonic guess works well, and that the stepwise SCF algorithm requires less computational time than the simultaneous SCF algorithm.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

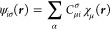


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


