{"title":"LARP7变异的功能特征:三例Alazami综合征新病例报告及文献综述","authors":"Anastasia Ambrose, Oana Caluseriu, Saadet Mercimek-Andrews","doi":"10.1155/humu/6490124","DOIUrl":null,"url":null,"abstract":"<p><b>Introduction:</b> Biallelic pathogenic variants in <i>LARP7</i> result in Alazami syndrome, which is characterized by global developmental delay, cognitive dysfunction, and dysmorphic features. Cardiac and skeletal phenotypes are reported in about 30% of individuals. We report three new individuals with Alazami syndrome and functional characterization of <i>LARP7</i> variants in this study.</p><p><b>Materials and Methods:</b> We reviewed electronic patient charts. We applied the American College of Medical Genetics and Genomics and the Association for Molecular Pathology variant classification algorithms. We performed a 3D protein modeling tool for in silico prediction and functional characterization of <i>LARP7</i> variants using qPCR gene expression experiments. We reviewed the medical literature for Alazami syndrome and <i>LARP7</i>.</p><p><b>Results:</b> We report three individuals from two unrelated families with characteristic phenotypes suggestive of Alazami syndrome. We identified a homozygous novel missense <i>LARP7</i> likely pathogenic variant (p.Asp54Val) in Family 1 and a homozygous novel pathogenic <i>LARP7</i> variant (p.Lys219Glu∗) in Family 2 using clinical exome sequencing. 3D protein modeling showed large structural changes for both variants compared to wildtype. The functional characterization showed a statistically significant difference in LARP7 expression between affected individuals and wildtype control. We report phenotypic variability within the same family that the cardiac phenotype was only present in Family 1, Case 2. There were < 60 individuals with Alazami syndrome reported to date.</p><p><b>Conclusion:</b> We report three new individuals with Alazami syndrome and two novel variants in <i>LARP7</i>. We report the first missense <i>LARP7</i> variant associated with Alazami syndrome. We report the protein 3D structure of <i>LARP7</i> variants. We show a relationship between the p.Asp54Val <i>LARP7</i> variant and LARP7 expression levels. We think that this could be due to abnormal RNA binding of LARP7 as per the 3D protein modeling prediction tool.</p>","PeriodicalId":13061,"journal":{"name":"Human Mutation","volume":"2025 1","pages":""},"PeriodicalIF":3.7000,"publicationDate":"2025-06-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1155/humu/6490124","citationCount":"0","resultStr":"{\"title\":\"Functional Characterization of Variants in LARP7: Report of Three New Individuals With Alazami Syndrome and a Literature Review\",\"authors\":\"Anastasia Ambrose, Oana Caluseriu, Saadet Mercimek-Andrews\",\"doi\":\"10.1155/humu/6490124\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><b>Introduction:</b> Biallelic pathogenic variants in <i>LARP7</i> result in Alazami syndrome, which is characterized by global developmental delay, cognitive dysfunction, and dysmorphic features. Cardiac and skeletal phenotypes are reported in about 30% of individuals. We report three new individuals with Alazami syndrome and functional characterization of <i>LARP7</i> variants in this study.</p><p><b>Materials and Methods:</b> We reviewed electronic patient charts. We applied the American College of Medical Genetics and Genomics and the Association for Molecular Pathology variant classification algorithms. We performed a 3D protein modeling tool for in silico prediction and functional characterization of <i>LARP7</i> variants using qPCR gene expression experiments. We reviewed the medical literature for Alazami syndrome and <i>LARP7</i>.</p><p><b>Results:</b> We report three individuals from two unrelated families with characteristic phenotypes suggestive of Alazami syndrome. We identified a homozygous novel missense <i>LARP7</i> likely pathogenic variant (p.Asp54Val) in Family 1 and a homozygous novel pathogenic <i>LARP7</i> variant (p.Lys219Glu∗) in Family 2 using clinical exome sequencing. 3D protein modeling showed large structural changes for both variants compared to wildtype. The functional characterization showed a statistically significant difference in LARP7 expression between affected individuals and wildtype control. We report phenotypic variability within the same family that the cardiac phenotype was only present in Family 1, Case 2. There were < 60 individuals with Alazami syndrome reported to date.</p><p><b>Conclusion:</b> We report three new individuals with Alazami syndrome and two novel variants in <i>LARP7</i>. We report the first missense <i>LARP7</i> variant associated with Alazami syndrome. We report the protein 3D structure of <i>LARP7</i> variants. We show a relationship between the p.Asp54Val <i>LARP7</i> variant and LARP7 expression levels. We think that this could be due to abnormal RNA binding of LARP7 as per the 3D protein modeling prediction tool.</p>\",\"PeriodicalId\":13061,\"journal\":{\"name\":\"Human Mutation\",\"volume\":\"2025 1\",\"pages\":\"\"},\"PeriodicalIF\":3.7000,\"publicationDate\":\"2025-06-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1155/humu/6490124\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Human Mutation\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1155/humu/6490124\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Mutation","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1155/humu/6490124","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Functional Characterization of Variants in LARP7: Report of Three New Individuals With Alazami Syndrome and a Literature Review
Introduction: Biallelic pathogenic variants in LARP7 result in Alazami syndrome, which is characterized by global developmental delay, cognitive dysfunction, and dysmorphic features. Cardiac and skeletal phenotypes are reported in about 30% of individuals. We report three new individuals with Alazami syndrome and functional characterization of LARP7 variants in this study.
Materials and Methods: We reviewed electronic patient charts. We applied the American College of Medical Genetics and Genomics and the Association for Molecular Pathology variant classification algorithms. We performed a 3D protein modeling tool for in silico prediction and functional characterization of LARP7 variants using qPCR gene expression experiments. We reviewed the medical literature for Alazami syndrome and LARP7.
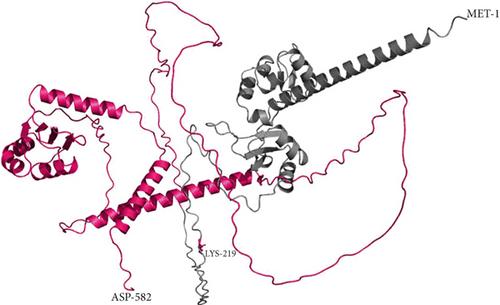
Results: We report three individuals from two unrelated families with characteristic phenotypes suggestive of Alazami syndrome. We identified a homozygous novel missense LARP7 likely pathogenic variant (p.Asp54Val) in Family 1 and a homozygous novel pathogenic LARP7 variant (p.Lys219Glu∗) in Family 2 using clinical exome sequencing. 3D protein modeling showed large structural changes for both variants compared to wildtype. The functional characterization showed a statistically significant difference in LARP7 expression between affected individuals and wildtype control. We report phenotypic variability within the same family that the cardiac phenotype was only present in Family 1, Case 2. There were < 60 individuals with Alazami syndrome reported to date.
Conclusion: We report three new individuals with Alazami syndrome and two novel variants in LARP7. We report the first missense LARP7 variant associated with Alazami syndrome. We report the protein 3D structure of LARP7 variants. We show a relationship between the p.Asp54Val LARP7 variant and LARP7 expression levels. We think that this could be due to abnormal RNA binding of LARP7 as per the 3D protein modeling prediction tool.
期刊介绍:
Human Mutation is a peer-reviewed journal that offers publication of original Research Articles, Methods, Mutation Updates, Reviews, Database Articles, Rapid Communications, and Letters on broad aspects of mutation research in humans. Reports of novel DNA variations and their phenotypic consequences, reports of SNPs demonstrated as valuable for genomic analysis, descriptions of new molecular detection methods, and novel approaches to clinical diagnosis are welcomed. Novel reports of gene organization at the genomic level, reported in the context of mutation investigation, may be considered. The journal provides a unique forum for the exchange of ideas, methods, and applications of interest to molecular, human, and medical geneticists in academic, industrial, and clinical research settings worldwide.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


