分子动力学模拟揭示了硫马锡及其水界面的结构和性质
IF 13.1
1区 工程技术
Q1 CONSTRUCTION & BUILDING TECHNOLOGY
引用次数: 0
摘要
硫镁石和钙矾石是造成硅酸盐水泥混凝土中硫酸盐侵蚀的主要原因。利用经典分子动力学(MD)计算机模拟技术,结合ClayFF-MOH力场,定量研究了土石的结构、振动、力学和热力学性质。此外,路径积分MD技术应用于更详细的检查结构和动力学的晶体内氢键在这种材料。经典MD模拟结果与现有实验数据和DFT计算结果吻合较好。模拟并比较了膨润土和钙矾石(0 0 0)和(0 0 0 1)表面的矿泉水界面。(0 0 0)和(0 0 0 1)膨润土和钙矾石表面的固水界面能为这些矿物的原子尺度溶解度机理提供了新的认识。模拟结果也证实了钙矾石表面有硫马石外延生长的可能性。本文章由计算机程序翻译,如有差异,请以英文原文为准。
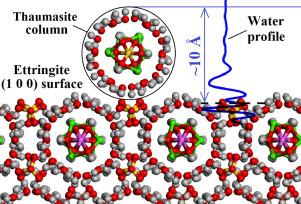
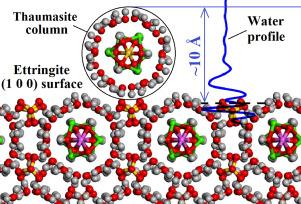
Structure and properties of thaumasite and its aqueous interfaces revealed by molecular dynamics simulations
Thaumasite, along with ettringite, is responsible for sulfate attack in concrete based on Portland cement. We use classical molecular dynamics (MD) computer simulation technique together with the ClayFF-MOH force field to quantitatively investigate the structural, vibrational, mechanical and thermodynamic properties of thaumasite. Additionally, path integral MD technique is applied for a more detailed examination of the structure and dynamics of intracrystalline hydrogen bonds in this material. The results of classical MD simulations are in good agreement with available experimental data and DFT calculations. Mineral-water interfaces for the (1 0 0) and (0 0 1) surfaces of thaumasite and ettringite are also simulated and compared. The solid-water interfacial energies for (1 0 0) and (0 0 1) thaumasite and ettringite surfaces provide new insights on the atomistic scale solubility mechanisms of these minerals. The simulations also confirm the possibility of thaumasite epitaxial growth on the surface of ettringite.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Cement and Concrete Research
工程技术-材料科学:综合
CiteScore
20.90
自引率
12.30%
发文量
318
审稿时长
53 days
期刊介绍:
Cement and Concrete Research is dedicated to publishing top-notch research on the materials science and engineering of cement, cement composites, mortars, concrete, and related materials incorporating cement or other mineral binders. The journal prioritizes reporting significant findings in research on the properties and performance of cementitious materials. It also covers novel experimental techniques, the latest analytical and modeling methods, examination and diagnosis of actual cement and concrete structures, and the exploration of potential improvements in materials.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


