Silabrata Pahari, Chi Ho Lee, Denis Johnson, David Kumar Yesudoss, Parth Shah, Mark A. Barteau, Abdoulaye Djire* and Joseph Sang-Il Kwon*,
{"title":"催化反应动力学研究进展:预测有效活化能的混合模型方法","authors":"Silabrata Pahari, Chi Ho Lee, Denis Johnson, David Kumar Yesudoss, Parth Shah, Mark A. Barteau, Abdoulaye Djire* and Joseph Sang-Il Kwon*, ","doi":"10.1021/acscatal.5c0207710.1021/acscatal.5c02077","DOIUrl":null,"url":null,"abstract":"<p >This study addresses limitations of traditional kinetic Monte Carlo (kMC) simulations, particularly their inability to capture latent surface dynamics on electrocatalysts due to complex many-body interactions among adsorbates, reactants, and intermediates. These shortcomings limit their predictive accuracy, often exacerbated by the separate limitations of density functional theory (DFT) in calculating activation energies (<i>E</i><sub>a</sub>) accurately. To overcome these challenges, we introduced a hybrid model that combines advanced machine learning (ML) with first-principles kMC. In this work, our approach defines “effective” activation energies incorporating nuanced physical phenomena that are absent in conventional kinetic models but evident in experiments. This hybrid model leverages these predictive effective activation energies to unveil underlying chemical phenomena occurring on catalyst surfaces. These phenomena include changes in product generation, surface coverage, and the dominant reaction mechanisms over time, which have been validated through experimental outcomes using MXene catalysts. Additionally, the ML component of our model not only provides an empirical fit but also infers underlying parameters that guide the subsequent DFT calculations based on changeable surface coverage over time.</p>","PeriodicalId":9,"journal":{"name":"ACS Catalysis ","volume":"15 11","pages":"9544–9554 9544–9554"},"PeriodicalIF":13.1000,"publicationDate":"2025-05-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Advancing Kinetic Study of Catalytic Reaction: A Hybrid Modeling Approach for Predicting Effective Activation Energies\",\"authors\":\"Silabrata Pahari, Chi Ho Lee, Denis Johnson, David Kumar Yesudoss, Parth Shah, Mark A. Barteau, Abdoulaye Djire* and Joseph Sang-Il Kwon*, \",\"doi\":\"10.1021/acscatal.5c0207710.1021/acscatal.5c02077\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >This study addresses limitations of traditional kinetic Monte Carlo (kMC) simulations, particularly their inability to capture latent surface dynamics on electrocatalysts due to complex many-body interactions among adsorbates, reactants, and intermediates. These shortcomings limit their predictive accuracy, often exacerbated by the separate limitations of density functional theory (DFT) in calculating activation energies (<i>E</i><sub>a</sub>) accurately. To overcome these challenges, we introduced a hybrid model that combines advanced machine learning (ML) with first-principles kMC. In this work, our approach defines “effective” activation energies incorporating nuanced physical phenomena that are absent in conventional kinetic models but evident in experiments. This hybrid model leverages these predictive effective activation energies to unveil underlying chemical phenomena occurring on catalyst surfaces. These phenomena include changes in product generation, surface coverage, and the dominant reaction mechanisms over time, which have been validated through experimental outcomes using MXene catalysts. Additionally, the ML component of our model not only provides an empirical fit but also infers underlying parameters that guide the subsequent DFT calculations based on changeable surface coverage over time.</p>\",\"PeriodicalId\":9,\"journal\":{\"name\":\"ACS Catalysis \",\"volume\":\"15 11\",\"pages\":\"9544–9554 9544–9554\"},\"PeriodicalIF\":13.1000,\"publicationDate\":\"2025-05-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ACS Catalysis \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acscatal.5c02077\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Catalysis ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acscatal.5c02077","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
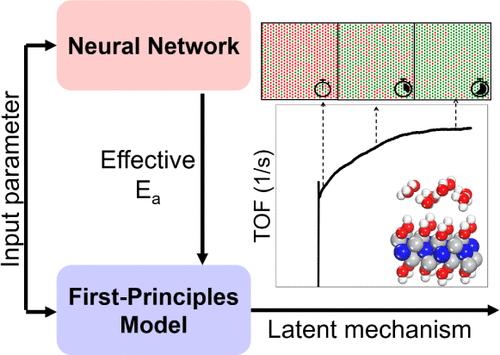
Advancing Kinetic Study of Catalytic Reaction: A Hybrid Modeling Approach for Predicting Effective Activation Energies
This study addresses limitations of traditional kinetic Monte Carlo (kMC) simulations, particularly their inability to capture latent surface dynamics on electrocatalysts due to complex many-body interactions among adsorbates, reactants, and intermediates. These shortcomings limit their predictive accuracy, often exacerbated by the separate limitations of density functional theory (DFT) in calculating activation energies (Ea) accurately. To overcome these challenges, we introduced a hybrid model that combines advanced machine learning (ML) with first-principles kMC. In this work, our approach defines “effective” activation energies incorporating nuanced physical phenomena that are absent in conventional kinetic models but evident in experiments. This hybrid model leverages these predictive effective activation energies to unveil underlying chemical phenomena occurring on catalyst surfaces. These phenomena include changes in product generation, surface coverage, and the dominant reaction mechanisms over time, which have been validated through experimental outcomes using MXene catalysts. Additionally, the ML component of our model not only provides an empirical fit but also infers underlying parameters that guide the subsequent DFT calculations based on changeable surface coverage over time.
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


