Desiree Henares, Meritxell Cubero, Irene Martinez-de-Albeniz, Alba Arranz, Muntsa Rocafort, Pedro Brotons, Amaresh Perez-Argüello, Maria Jose Troyano, Amadeu Gene, Aleix Lluansi, Marti Iriondo-Sanz, Iolanda Jordan, Claudia Fortuny, Mireia Urrea, C Muñoz-Almagro
{"title":"通过第三代长读纳米孔测序快速识别新生儿重症监护病房的粘质沙雷氏菌暴发。","authors":"Desiree Henares, Meritxell Cubero, Irene Martinez-de-Albeniz, Alba Arranz, Muntsa Rocafort, Pedro Brotons, Amaresh Perez-Argüello, Maria Jose Troyano, Amadeu Gene, Aleix Lluansi, Marti Iriondo-Sanz, Iolanda Jordan, Claudia Fortuny, Mireia Urrea, C Muñoz-Almagro","doi":"10.1186/s13756-025-01582-x","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Serratia marcescens is a frequent cause of outbreaks in high-risk hospital settings such as neonatal intensive care units (NICU). This study investigated a potential S. marcescens outbreak in the NICU of a reference children's hospital using Whole Genome Sequencing (WGS). Additionally, it assessed the performance of third-generation sequencing for the rapid and accurate identification and characterization of the outbreak's clonal strain.</p><p><strong>Methods: </strong>A prospective study was conducted from September 8th to November 12th 2021, following a sharp increase in invasive S. marcescens infections in the NICU of University Children's Hospital Sant Joan de Déu (Barcelona, Spain). This study included all patients admitted to NICU and other hospital wards from whom S. marcescens was isolated in any sample type. Nanopore sequencing was performed on S. marcescens isolates. Genomic characterization included phylogenetic analyses and detection of antimicrobial resistance genes.</p><p><strong>Results: </strong>Twenty-nine patients (16 NICU and 13 non-NICU patients) infected/colonized by S. marcescens were detected during the study period, accounting for a total of 61 isolates. The genomic characterization was performed on 24 isolates from 14 NICU-patients and 10 isolates from eight non-NICU patients. Phylogenetic analyses evidenced three clusters of closely related strains; cluster I (n = 22), II (n = 2) and III (n = 5). The remaining isolates (n = 5) did not cluster. Cluster I contained most isolates from NICU patients (20/24), and most isolates from NICU-patients with confirmed invasive disease (7/8). Cluster II contained two isolates from two NICU-patients, one presenting with invasive disease. The resistance gene blaSRT was found in 97% of S. marcescens isolates (33/34). All isolates exhibited the amikacin-tobramycin aac(6') resistance gene and three multi-drug efflux pumps genes; sdeY, sdeB and smfY. The tetracycline tet(41) resistance gene was found in non-clustered isolates (4/34). The first results were available less than one month after the outbreak's alarm, and complete genomic study after two months.</p><p><strong>Conclusion: </strong>Two clonal strains were co-circulating in the NICU setting, with one being the major strain responsible for the outbreak. Rapid molecular characterization with nanopore sequencing confirmed the outbreak. It revealed the phylogenetic relationships among isolates and their antimicrobial potential. This approach enabled effective contextualization of the outbreak and allowed for monitoring its progression.</p>","PeriodicalId":7950,"journal":{"name":"Antimicrobial Resistance and Infection Control","volume":"14 1","pages":"63"},"PeriodicalIF":4.4000,"publicationDate":"2025-06-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12135216/pdf/","citationCount":"0","resultStr":"{\"title\":\"Rapid identification of a Serratia marcescens outbreak in a neonatal intensive care unit by third-generation long-read nanopore sequencing.\",\"authors\":\"Desiree Henares, Meritxell Cubero, Irene Martinez-de-Albeniz, Alba Arranz, Muntsa Rocafort, Pedro Brotons, Amaresh Perez-Argüello, Maria Jose Troyano, Amadeu Gene, Aleix Lluansi, Marti Iriondo-Sanz, Iolanda Jordan, Claudia Fortuny, Mireia Urrea, C Muñoz-Almagro\",\"doi\":\"10.1186/s13756-025-01582-x\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Serratia marcescens is a frequent cause of outbreaks in high-risk hospital settings such as neonatal intensive care units (NICU). This study investigated a potential S. marcescens outbreak in the NICU of a reference children's hospital using Whole Genome Sequencing (WGS). Additionally, it assessed the performance of third-generation sequencing for the rapid and accurate identification and characterization of the outbreak's clonal strain.</p><p><strong>Methods: </strong>A prospective study was conducted from September 8th to November 12th 2021, following a sharp increase in invasive S. marcescens infections in the NICU of University Children's Hospital Sant Joan de Déu (Barcelona, Spain). This study included all patients admitted to NICU and other hospital wards from whom S. marcescens was isolated in any sample type. Nanopore sequencing was performed on S. marcescens isolates. Genomic characterization included phylogenetic analyses and detection of antimicrobial resistance genes.</p><p><strong>Results: </strong>Twenty-nine patients (16 NICU and 13 non-NICU patients) infected/colonized by S. marcescens were detected during the study period, accounting for a total of 61 isolates. The genomic characterization was performed on 24 isolates from 14 NICU-patients and 10 isolates from eight non-NICU patients. Phylogenetic analyses evidenced three clusters of closely related strains; cluster I (n = 22), II (n = 2) and III (n = 5). The remaining isolates (n = 5) did not cluster. Cluster I contained most isolates from NICU patients (20/24), and most isolates from NICU-patients with confirmed invasive disease (7/8). Cluster II contained two isolates from two NICU-patients, one presenting with invasive disease. The resistance gene blaSRT was found in 97% of S. marcescens isolates (33/34). All isolates exhibited the amikacin-tobramycin aac(6') resistance gene and three multi-drug efflux pumps genes; sdeY, sdeB and smfY. The tetracycline tet(41) resistance gene was found in non-clustered isolates (4/34). The first results were available less than one month after the outbreak's alarm, and complete genomic study after two months.</p><p><strong>Conclusion: </strong>Two clonal strains were co-circulating in the NICU setting, with one being the major strain responsible for the outbreak. Rapid molecular characterization with nanopore sequencing confirmed the outbreak. It revealed the phylogenetic relationships among isolates and their antimicrobial potential. This approach enabled effective contextualization of the outbreak and allowed for monitoring its progression.</p>\",\"PeriodicalId\":7950,\"journal\":{\"name\":\"Antimicrobial Resistance and Infection Control\",\"volume\":\"14 1\",\"pages\":\"63\"},\"PeriodicalIF\":4.4000,\"publicationDate\":\"2025-06-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12135216/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Antimicrobial Resistance and Infection Control\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13756-025-01582-x\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"INFECTIOUS DISEASES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Antimicrobial Resistance and Infection Control","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13756-025-01582-x","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"INFECTIOUS DISEASES","Score":null,"Total":0}
引用次数: 0
摘要
背景:粘质沙雷菌是高风险医院环境(如新生儿重症监护病房(NICU))暴发的常见原因。本研究利用全基因组测序(WGS)调查了某参考儿童医院NICU中潜在的粘质葡萄球菌暴发。此外,它还评估了第三代测序在快速准确识别和表征暴发克隆菌株方面的性能。方法:2021年9月8日至11月12日,在西班牙巴塞罗那圣胡安儿童医院(Sant Joan de dassau) NICU侵袭性粘质链球菌感染急剧增加之后,进行了一项前瞻性研究。本研究包括所有在NICU和其他医院病房中分离出粘质葡萄球菌的患者。对粘质葡萄球菌分离物进行纳米孔测序。基因组鉴定包括系统发育分析和耐药基因检测。结果:研究期间共检出粘质葡萄球菌感染/定植患者29例(NICU 16例,非NICU 13例),共分离61株。对来自14例nicu患者的24株分离株和来自8例非nicu患者的10株分离株进行基因组鉴定。系统发育分析证实了三个密切相关的菌株群;集群I (n = 22),集群II (n = 2)和集群III (n = 5)。其余分离株(n = 5)未聚集。聚类I中来自NICU患者的分离株最多(20/24),来自确诊有侵袭性疾病的NICU患者的分离株最多(7/8)。集群II包含来自两名新生儿重症监护病房患者的两株分离株,其中一株表现为侵袭性疾病。耐药基因blaSRT在97%的粘质葡萄球菌分离株中被发现(33/34)。所有分离株均具有阿米卡霉素-妥布霉素aac(6’)耐药基因和3个多药外排泵基因;sdeY, sdeB和smfY。在非聚集性分离株中发现四环素tet(41)耐药基因(4/34)。第一批结果在疫情警报发出不到一个月后出炉,完整的基因组研究在两个月后出炉。结论:两株克隆菌株在NICU环境中共流行,其中一株是导致暴发的主要菌株。采用纳米孔测序技术的快速分子表征证实了此次疫情。揭示了分离株的系统发育关系及其抗菌潜力。这种方法能够有效地对疫情进行背景分析,并能够监测其进展。
Rapid identification of a Serratia marcescens outbreak in a neonatal intensive care unit by third-generation long-read nanopore sequencing.
Background: Serratia marcescens is a frequent cause of outbreaks in high-risk hospital settings such as neonatal intensive care units (NICU). This study investigated a potential S. marcescens outbreak in the NICU of a reference children's hospital using Whole Genome Sequencing (WGS). Additionally, it assessed the performance of third-generation sequencing for the rapid and accurate identification and characterization of the outbreak's clonal strain.
Methods: A prospective study was conducted from September 8th to November 12th 2021, following a sharp increase in invasive S. marcescens infections in the NICU of University Children's Hospital Sant Joan de Déu (Barcelona, Spain). This study included all patients admitted to NICU and other hospital wards from whom S. marcescens was isolated in any sample type. Nanopore sequencing was performed on S. marcescens isolates. Genomic characterization included phylogenetic analyses and detection of antimicrobial resistance genes.
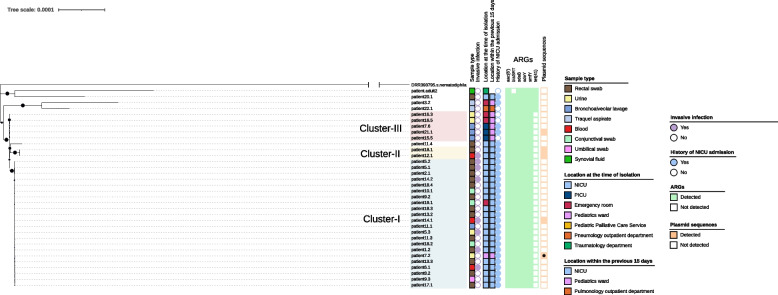
Results: Twenty-nine patients (16 NICU and 13 non-NICU patients) infected/colonized by S. marcescens were detected during the study period, accounting for a total of 61 isolates. The genomic characterization was performed on 24 isolates from 14 NICU-patients and 10 isolates from eight non-NICU patients. Phylogenetic analyses evidenced three clusters of closely related strains; cluster I (n = 22), II (n = 2) and III (n = 5). The remaining isolates (n = 5) did not cluster. Cluster I contained most isolates from NICU patients (20/24), and most isolates from NICU-patients with confirmed invasive disease (7/8). Cluster II contained two isolates from two NICU-patients, one presenting with invasive disease. The resistance gene blaSRT was found in 97% of S. marcescens isolates (33/34). All isolates exhibited the amikacin-tobramycin aac(6') resistance gene and three multi-drug efflux pumps genes; sdeY, sdeB and smfY. The tetracycline tet(41) resistance gene was found in non-clustered isolates (4/34). The first results were available less than one month after the outbreak's alarm, and complete genomic study after two months.
Conclusion: Two clonal strains were co-circulating in the NICU setting, with one being the major strain responsible for the outbreak. Rapid molecular characterization with nanopore sequencing confirmed the outbreak. It revealed the phylogenetic relationships among isolates and their antimicrobial potential. This approach enabled effective contextualization of the outbreak and allowed for monitoring its progression.
期刊介绍:
Antimicrobial Resistance and Infection Control is a global forum for all those working on the prevention, diagnostic and treatment of health-care associated infections and antimicrobial resistance development in all health-care settings. The journal covers a broad spectrum of preeminent practices and best available data to the top interventional and translational research, and innovative developments in the field of infection control.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


