Eiichi Tokuda, Laura Leykam, Per Zetterström, Thomas Brännström, Peter M Andersen, Stefan L Marklund
{"title":"人类SOD1变异共表达对运动神经元疾病的多种影响。","authors":"Eiichi Tokuda, Laura Leykam, Per Zetterström, Thomas Brännström, Peter M Andersen, Stefan L Marklund","doi":"10.1093/hmg/ddaf088","DOIUrl":null,"url":null,"abstract":"<p><p>Mutations in superoxide dismutase-1 (SOD1) are a common cause of amyotrophic lateral sclerosis (ALS). Inheritance is as a rule dominant, but in carriers of the most common mutation, D90A, disease can develop in both homozygous and, more rarely, in heterozygous individuals with unexplained differences in clinical presentation. There is mounting evidence that prion-like spread of SOD1 aggregation is the primary cause of the disease. Two different strains of aggregates have been found to arise in human SOD1 (hSOD1) transgenic mouse models of ALS. Strain A is formed by most mutants including hSOD1G85R and hSOD1WT, whereas hSOD1D90A transgenic mice form a distinct strain B in addition to A. To explore the effects of aggregate strain propensities when hSOD1 variants are coexpressed, we generated digenic hSOD1G85R/WT and hSOD1G85R/D90A mice. Coexpression of hSOD1WT considerably shortened the lifespan of hSOD1G85R mice to the extent expected from the neurotoxicities of the variants alone. In contrast, coexpression of hSOD1D90A had a minimal effect on survival, far smaller than expected. Moreover, time from onset to the end stage was markedly prolonged in the hSOD1G85R/D90A mice. Aggregation of hSOD1 developed concomitantly with motor neuron disease, and the aggregates contained large amounts of both coexpressed variants in both digenic models. Our findings suggest that hSOD1WT has high a capacity to coaggregate with mutants and enhance neurotoxicity. Such interactions may be restricted by differences in strain propensities, which may contribute to the primarily recessive inheritance associated with the hSOD1D90A mutation.</p>","PeriodicalId":13070,"journal":{"name":"Human molecular genetics","volume":" ","pages":"1380-1391"},"PeriodicalIF":3.2000,"publicationDate":"2025-08-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12361113/pdf/","citationCount":"0","resultStr":"{\"title\":\"Diverse effects of coexpression of human SOD1 variants on motor neuron disease.\",\"authors\":\"Eiichi Tokuda, Laura Leykam, Per Zetterström, Thomas Brännström, Peter M Andersen, Stefan L Marklund\",\"doi\":\"10.1093/hmg/ddaf088\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Mutations in superoxide dismutase-1 (SOD1) are a common cause of amyotrophic lateral sclerosis (ALS). Inheritance is as a rule dominant, but in carriers of the most common mutation, D90A, disease can develop in both homozygous and, more rarely, in heterozygous individuals with unexplained differences in clinical presentation. There is mounting evidence that prion-like spread of SOD1 aggregation is the primary cause of the disease. Two different strains of aggregates have been found to arise in human SOD1 (hSOD1) transgenic mouse models of ALS. Strain A is formed by most mutants including hSOD1G85R and hSOD1WT, whereas hSOD1D90A transgenic mice form a distinct strain B in addition to A. To explore the effects of aggregate strain propensities when hSOD1 variants are coexpressed, we generated digenic hSOD1G85R/WT and hSOD1G85R/D90A mice. Coexpression of hSOD1WT considerably shortened the lifespan of hSOD1G85R mice to the extent expected from the neurotoxicities of the variants alone. In contrast, coexpression of hSOD1D90A had a minimal effect on survival, far smaller than expected. Moreover, time from onset to the end stage was markedly prolonged in the hSOD1G85R/D90A mice. Aggregation of hSOD1 developed concomitantly with motor neuron disease, and the aggregates contained large amounts of both coexpressed variants in both digenic models. Our findings suggest that hSOD1WT has high a capacity to coaggregate with mutants and enhance neurotoxicity. Such interactions may be restricted by differences in strain propensities, which may contribute to the primarily recessive inheritance associated with the hSOD1D90A mutation.</p>\",\"PeriodicalId\":13070,\"journal\":{\"name\":\"Human molecular genetics\",\"volume\":\" \",\"pages\":\"1380-1391\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2025-08-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12361113/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Human molecular genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/hmg/ddaf088\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human molecular genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/hmg/ddaf088","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Diverse effects of coexpression of human SOD1 variants on motor neuron disease.
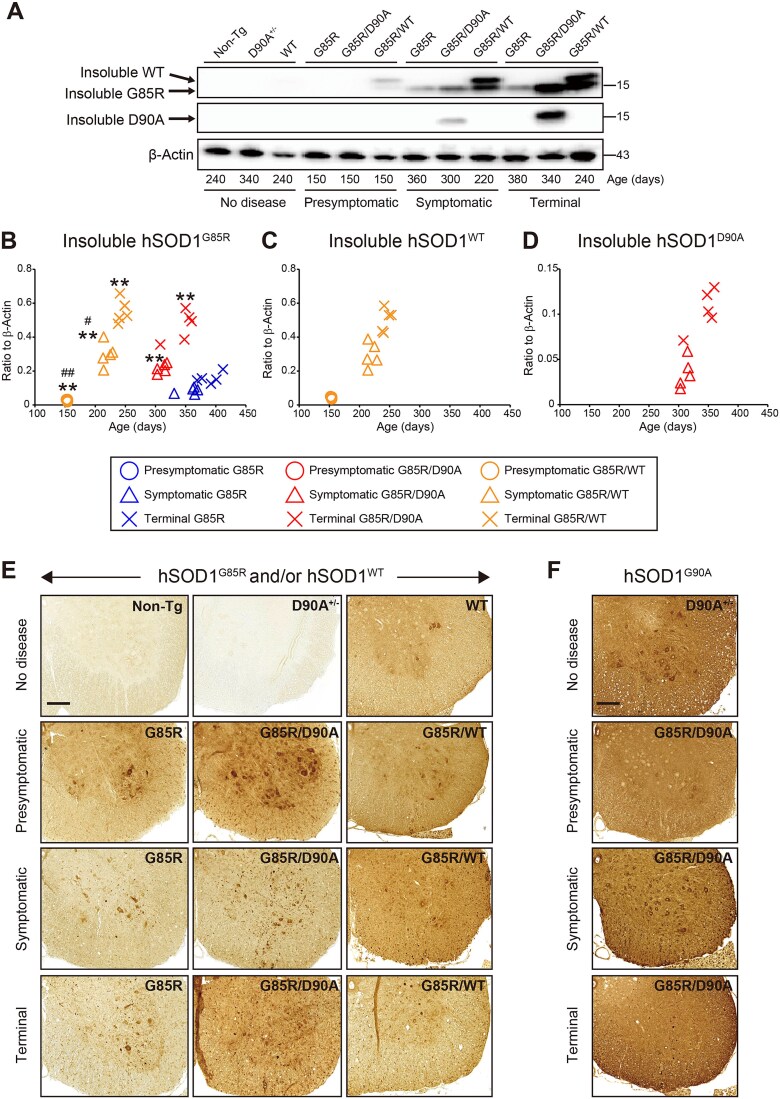
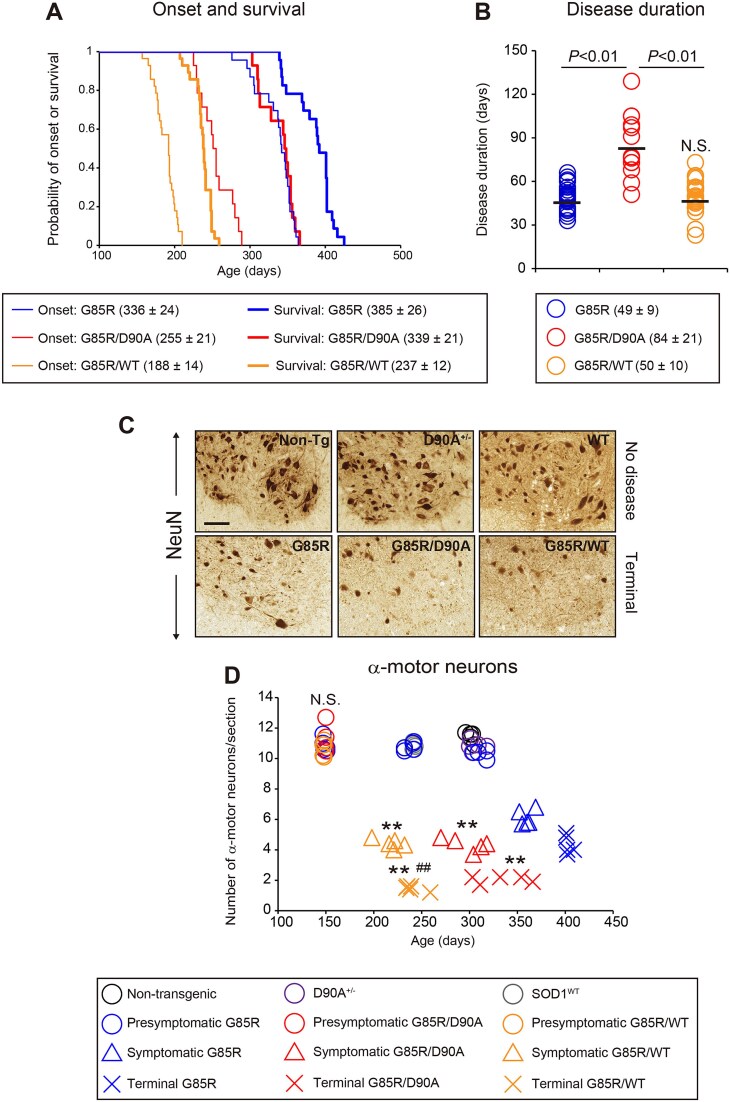
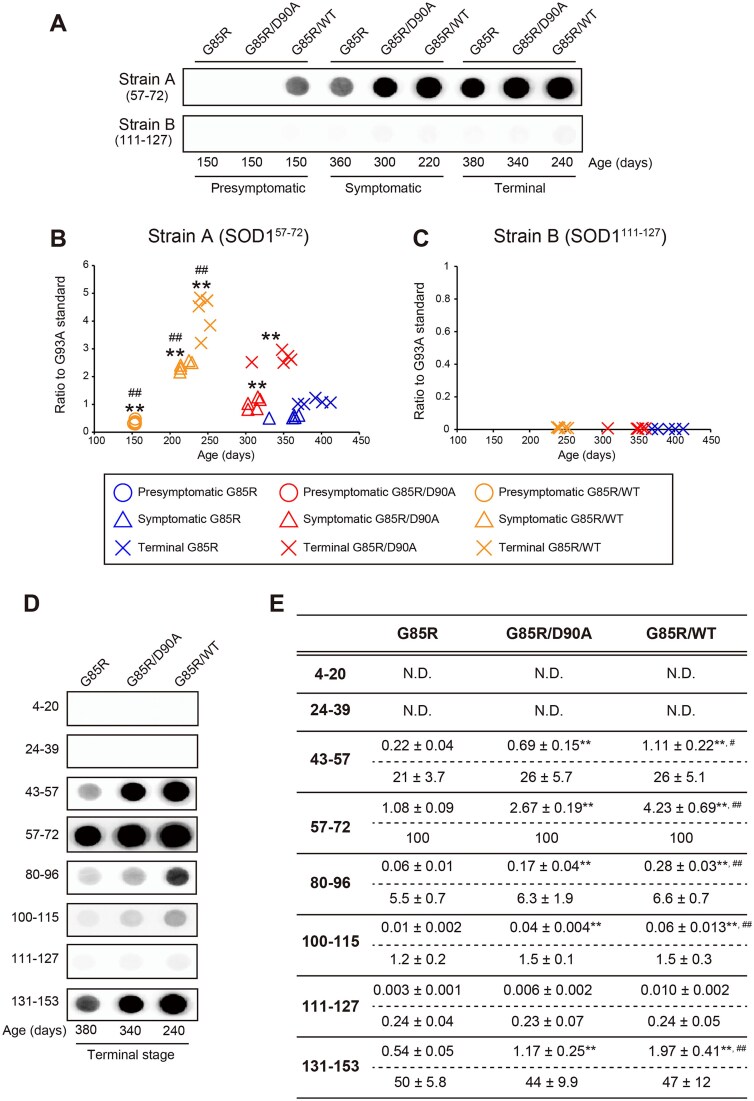
Mutations in superoxide dismutase-1 (SOD1) are a common cause of amyotrophic lateral sclerosis (ALS). Inheritance is as a rule dominant, but in carriers of the most common mutation, D90A, disease can develop in both homozygous and, more rarely, in heterozygous individuals with unexplained differences in clinical presentation. There is mounting evidence that prion-like spread of SOD1 aggregation is the primary cause of the disease. Two different strains of aggregates have been found to arise in human SOD1 (hSOD1) transgenic mouse models of ALS. Strain A is formed by most mutants including hSOD1G85R and hSOD1WT, whereas hSOD1D90A transgenic mice form a distinct strain B in addition to A. To explore the effects of aggregate strain propensities when hSOD1 variants are coexpressed, we generated digenic hSOD1G85R/WT and hSOD1G85R/D90A mice. Coexpression of hSOD1WT considerably shortened the lifespan of hSOD1G85R mice to the extent expected from the neurotoxicities of the variants alone. In contrast, coexpression of hSOD1D90A had a minimal effect on survival, far smaller than expected. Moreover, time from onset to the end stage was markedly prolonged in the hSOD1G85R/D90A mice. Aggregation of hSOD1 developed concomitantly with motor neuron disease, and the aggregates contained large amounts of both coexpressed variants in both digenic models. Our findings suggest that hSOD1WT has high a capacity to coaggregate with mutants and enhance neurotoxicity. Such interactions may be restricted by differences in strain propensities, which may contribute to the primarily recessive inheritance associated with the hSOD1D90A mutation.
期刊介绍:
Human Molecular Genetics concentrates on full-length research papers covering a wide range of topics in all aspects of human molecular genetics. These include:
the molecular basis of human genetic disease
developmental genetics
cancer genetics
neurogenetics
chromosome and genome structure and function
therapy of genetic disease
stem cells in human genetic disease and therapy, including the application of iPS cells
genome-wide association studies
mouse and other models of human diseases
functional genomics
computational genomics
In addition, the journal also publishes research on other model systems for the analysis of genes, especially when there is an obvious relevance to human genetics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


