Elaine Zhang, Teresa Zhao, Tim Sikora, Carolyn Ellaway, Wendy A. Gold, Nicole J. Van Bergen, David A. Stroud, John Christodoulou, Simranpreet Kaur
{"title":"CHD8变异和Rett综合征:重叠表型、分子趋同和扩大遗传谱","authors":"Elaine Zhang, Teresa Zhao, Tim Sikora, Carolyn Ellaway, Wendy A. Gold, Nicole J. Van Bergen, David A. Stroud, John Christodoulou, Simranpreet Kaur","doi":"10.1155/humu/5485987","DOIUrl":null,"url":null,"abstract":"<p>Rett syndrome (RTT) is a rare, X-linked, severe neurodevelopmental disorder, predominantly associated with pathogenic variants in the methyl-CpG-binding protein-2 (<i>MECP2</i>) gene, with an increasing number of atypical RTT or RTT-like individuals having pathogenic variants in other genes, such as cyclin-dependent kinase-like 5 (<i>CDKL5</i>) or forkhead box G1 (<i>FOXG1</i>). However, ~20% of individuals with a clinical diagnosis of RTT remain genetically undiagnosed, highlighting the importance of ongoing genomic and functional studies to expand the genetic spectrum of RTT. We present a female who was born to healthy nonconsanguineous parents and presented with severe intellectual disability, macrocephaly, ataxia, absent speech, and poor eye contact. The affected individual was clinically diagnosed with atypical RTT, but genetic testing showed no pathogenic variants in <i>MECP2</i>, <i>CDKL5</i>, or <i>FOXG1.</i> Singleton whole genome sequencing was conducted, which identified a heterozygous stop–gain variant [NM_001170629.2: c.5017C>T, p.(Arg1673 <sup>∗</sup>)], in the chromodomain-helicase-DNA-binding protein 8 (<i>CHD8</i>) gene. Variant curation revealed its absence in unaffected populations, in silico predictions of pathogenicity, and an existing association with <i>intellectual developmental disorder with autism and macrocephaly</i> (<i>IDDAM</i>) (OMIM #615032). In vitro functional analyses, including Western blots, quantitative reverse transcription polymerase chain reaction (qRT-PCR), and proteomic analyses, demonstrated a significant reduction of the CHD8 transcript and two CHD8 protein isoforms in the proband’s skin fibroblasts relative to control fibroblasts. Additionally, proteomic analysis indicated a significant reduction of the MeCP2 protein, indicating a possible molecular link between CHD8 and MeCP2 and thus clinically between IDDAM and RTT. As the affected individual’s phenotype is consistent with atypical RTT, our results suggest that <i>CHD8</i> could be considered in the expanding genetic spectrum of atypical RTT, which may assist the diagnosis of other <i>MECP2</i>-negative RTT individuals.</p>","PeriodicalId":13061,"journal":{"name":"Human Mutation","volume":"2025 1","pages":""},"PeriodicalIF":3.7000,"publicationDate":"2025-06-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1155/humu/5485987","citationCount":"0","resultStr":"{\"title\":\"CHD8 Variant and Rett Syndrome: Overlapping Phenotypes, Molecular Convergence, and Expanding the Genetic Spectrum\",\"authors\":\"Elaine Zhang, Teresa Zhao, Tim Sikora, Carolyn Ellaway, Wendy A. Gold, Nicole J. Van Bergen, David A. Stroud, John Christodoulou, Simranpreet Kaur\",\"doi\":\"10.1155/humu/5485987\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Rett syndrome (RTT) is a rare, X-linked, severe neurodevelopmental disorder, predominantly associated with pathogenic variants in the methyl-CpG-binding protein-2 (<i>MECP2</i>) gene, with an increasing number of atypical RTT or RTT-like individuals having pathogenic variants in other genes, such as cyclin-dependent kinase-like 5 (<i>CDKL5</i>) or forkhead box G1 (<i>FOXG1</i>). However, ~20% of individuals with a clinical diagnosis of RTT remain genetically undiagnosed, highlighting the importance of ongoing genomic and functional studies to expand the genetic spectrum of RTT. We present a female who was born to healthy nonconsanguineous parents and presented with severe intellectual disability, macrocephaly, ataxia, absent speech, and poor eye contact. The affected individual was clinically diagnosed with atypical RTT, but genetic testing showed no pathogenic variants in <i>MECP2</i>, <i>CDKL5</i>, or <i>FOXG1.</i> Singleton whole genome sequencing was conducted, which identified a heterozygous stop–gain variant [NM_001170629.2: c.5017C>T, p.(Arg1673 <sup>∗</sup>)], in the chromodomain-helicase-DNA-binding protein 8 (<i>CHD8</i>) gene. Variant curation revealed its absence in unaffected populations, in silico predictions of pathogenicity, and an existing association with <i>intellectual developmental disorder with autism and macrocephaly</i> (<i>IDDAM</i>) (OMIM #615032). In vitro functional analyses, including Western blots, quantitative reverse transcription polymerase chain reaction (qRT-PCR), and proteomic analyses, demonstrated a significant reduction of the CHD8 transcript and two CHD8 protein isoforms in the proband’s skin fibroblasts relative to control fibroblasts. Additionally, proteomic analysis indicated a significant reduction of the MeCP2 protein, indicating a possible molecular link between CHD8 and MeCP2 and thus clinically between IDDAM and RTT. As the affected individual’s phenotype is consistent with atypical RTT, our results suggest that <i>CHD8</i> could be considered in the expanding genetic spectrum of atypical RTT, which may assist the diagnosis of other <i>MECP2</i>-negative RTT individuals.</p>\",\"PeriodicalId\":13061,\"journal\":{\"name\":\"Human Mutation\",\"volume\":\"2025 1\",\"pages\":\"\"},\"PeriodicalIF\":3.7000,\"publicationDate\":\"2025-06-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1155/humu/5485987\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Human Mutation\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1155/humu/5485987\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Mutation","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1155/humu/5485987","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
CHD8 Variant and Rett Syndrome: Overlapping Phenotypes, Molecular Convergence, and Expanding the Genetic Spectrum
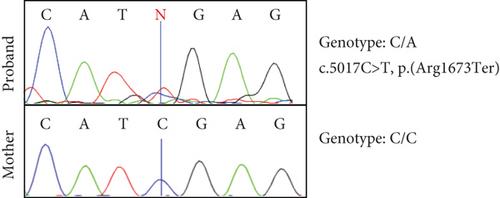
Rett syndrome (RTT) is a rare, X-linked, severe neurodevelopmental disorder, predominantly associated with pathogenic variants in the methyl-CpG-binding protein-2 (MECP2) gene, with an increasing number of atypical RTT or RTT-like individuals having pathogenic variants in other genes, such as cyclin-dependent kinase-like 5 (CDKL5) or forkhead box G1 (FOXG1). However, ~20% of individuals with a clinical diagnosis of RTT remain genetically undiagnosed, highlighting the importance of ongoing genomic and functional studies to expand the genetic spectrum of RTT. We present a female who was born to healthy nonconsanguineous parents and presented with severe intellectual disability, macrocephaly, ataxia, absent speech, and poor eye contact. The affected individual was clinically diagnosed with atypical RTT, but genetic testing showed no pathogenic variants in MECP2, CDKL5, or FOXG1. Singleton whole genome sequencing was conducted, which identified a heterozygous stop–gain variant [NM_001170629.2: c.5017C>T, p.(Arg1673 ∗)], in the chromodomain-helicase-DNA-binding protein 8 (CHD8) gene. Variant curation revealed its absence in unaffected populations, in silico predictions of pathogenicity, and an existing association with intellectual developmental disorder with autism and macrocephaly (IDDAM) (OMIM #615032). In vitro functional analyses, including Western blots, quantitative reverse transcription polymerase chain reaction (qRT-PCR), and proteomic analyses, demonstrated a significant reduction of the CHD8 transcript and two CHD8 protein isoforms in the proband’s skin fibroblasts relative to control fibroblasts. Additionally, proteomic analysis indicated a significant reduction of the MeCP2 protein, indicating a possible molecular link between CHD8 and MeCP2 and thus clinically between IDDAM and RTT. As the affected individual’s phenotype is consistent with atypical RTT, our results suggest that CHD8 could be considered in the expanding genetic spectrum of atypical RTT, which may assist the diagnosis of other MECP2-negative RTT individuals.
期刊介绍:
Human Mutation is a peer-reviewed journal that offers publication of original Research Articles, Methods, Mutation Updates, Reviews, Database Articles, Rapid Communications, and Letters on broad aspects of mutation research in humans. Reports of novel DNA variations and their phenotypic consequences, reports of SNPs demonstrated as valuable for genomic analysis, descriptions of new molecular detection methods, and novel approaches to clinical diagnosis are welcomed. Novel reports of gene organization at the genomic level, reported in the context of mutation investigation, may be considered. The journal provides a unique forum for the exchange of ideas, methods, and applications of interest to molecular, human, and medical geneticists in academic, industrial, and clinical research settings worldwide.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


