利用高效Gillespie算法模拟复杂网络上的疫情峰值动态
IF 2.6
4区 医学
Q3 INFECTIOUS DISEASES
引用次数: 0
摘要
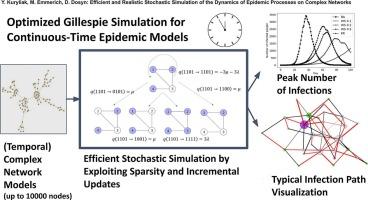
我们提出了一项流行病在复杂网络上传播的综合研究,(i)揭示了网络结构和有针对性的干预措施如何影响感染节点的峰值计数(PCIN)及其时间,(ii)提供了一个开源仪表板,让研究人员通过现实的模型扩展来探索这些影响。(iii)提供一种高性能仿真引擎,该引擎通过乘法因子提高Gillespie算法的现有稀疏网络实现的时间复杂度。连续时间SI/SIS/SIR动态分析了两个干预参数:边缘特异性感染率降低和节点级恢复加速,得出了跨异构拓扑的峰值高度和延迟的明确界限。为了交互式地测试场景,我们扩展了仪表板模拟器,以包括非指数恢复时间、时间重新布线、加权和多类型接触、抗原等效突变株的模拟以及可能感染途径的新型视觉聚合。此外,我们重新设计了Gillespie的稀疏图算法,其核心是通过简洁渐进地更新(和)转移率和维护排序的受感染节点列表,与最先进的邻接表实现相比,速度增加了一个乘法因子。当感染处于早期阶段且大型网络中只有少数节点被感染时,我们的新算法可以获得额外的速度增益;一个互补的密集矩阵变体涵盖了非稀疏情况。Barabási-Albert网络上的基准测试证实,与Gillespie算法的标准邻接表实现相比,该算法的增益达到了数量级。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Simulating epidemic peak dynamics on complex networks using efficient Gillespie algorithms
We present an integrated study of epidemic spreading on complex networks that (i) reveals how network structure and targeted interventions shape the peak count of infected nodes (PCIN) and its timing, (ii) supplies an open-source dashboard that lets researchers explore these effects with realistic model extensions, and (iii) delivers a high-performance simulation engine which improves the time complexity of existing sparse network implementations of Gillespie's algorithm by multiplicative factors. Continuous-time SI/SIS/SIR dynamics are analyzed with respect to two intervention knobs: edge-specific infection rate reduction and node-level recovery acceleration, yielding explicit bounds on peak height and delay across heterogeneous topologies. To test scenarios interactively, we extend the dashboard simulator to include non-exponential recovery times, temporal rewiring, weighted and multi-type contacts, simulation of antigenically equivalent mutant strains, and a novel visual aggregation of likely infection routes. Moreover, we redesign Gillespie's algorithm for sparse graphs at its core by succinctly and incrementally updating the (sums of the) transition rate and maintaining a sorted infected node list, achieving speed increases with a multiplicative factor compared to the state-of-the-art implementation of the adjacency list. Additional speed gains can be achieved by our new algorithm when infection is in its early stage and only a few nodes of a large network are infected; a complementary dense matrix variant covers non-sparse cases. Benchmarks on Barabási–Albert networks confirm up-to-order-of-magnitude gains over the standard adjacency-list implementation of Gillespie's algorithm.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Infection Genetics and Evolution
医学-传染病学
CiteScore
8.40
自引率
0.00%
发文量
215
审稿时长
82 days
期刊介绍:
(aka Journal of Molecular Epidemiology and Evolutionary Genetics of Infectious Diseases -- MEEGID)
Infectious diseases constitute one of the main challenges to medical science in the coming century. The impressive development of molecular megatechnologies and of bioinformatics have greatly increased our knowledge of the evolution, transmission and pathogenicity of infectious diseases. Research has shown that host susceptibility to many infectious diseases has a genetic basis. Furthermore, much is now known on the molecular epidemiology, evolution and virulence of pathogenic agents, as well as their resistance to drugs, vaccines, and antibiotics. Equally, research on the genetics of disease vectors has greatly improved our understanding of their systematics, has increased our capacity to identify target populations for control or intervention, and has provided detailed information on the mechanisms of insecticide resistance.
However, the genetics and evolutionary biology of hosts, pathogens and vectors have tended to develop as three separate fields of research. This artificial compartmentalisation is of concern due to our growing appreciation of the strong co-evolutionary interactions among hosts, pathogens and vectors.
Infection, Genetics and Evolution and its companion congress [MEEGID](http://www.meegidconference.com/) (for Molecular Epidemiology and Evolutionary Genetics of Infectious Diseases) are the main forum acting for the cross-fertilization between evolutionary science and biomedical research on infectious diseases.
Infection, Genetics and Evolution is the only journal that welcomes articles dealing with the genetics and evolutionary biology of hosts, pathogens and vectors, and coevolution processes among them in relation to infection and disease manifestation. All infectious models enter the scope of the journal, including pathogens of humans, animals and plants, either parasites, fungi, bacteria, viruses or prions. The journal welcomes articles dealing with genetics, population genetics, genomics, postgenomics, gene expression, evolutionary biology, population dynamics, mathematical modeling and bioinformatics. We also provide many author benefits, such as free PDFs, a liberal copyright policy, special discounts on Elsevier publications and much more. Please click here for more information on our author services .
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


