Krystallenia I Alexandraki, Odysseas Violetis, Eleni Memi, Helen Fryssira, Vasileios Papanikolaou, Maria Papagianni, George Mastorakos
{"title":"FGFR1变异女性罕见的促性腺功能减退、生长激素缺乏和直肠闭锁的合并:病例报告和文献系统回顾。","authors":"Krystallenia I Alexandraki, Odysseas Violetis, Eleni Memi, Helen Fryssira, Vasileios Papanikolaou, Maria Papagianni, George Mastorakos","doi":"10.1007/s12020-025-04261-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Purpose: </strong>To report a case with combined pituitary hormone deficiency (CPHD) and Fibroblast growth factor receptor 1 (FGFR1) gene defect, and summarize the clinical characteristics of similar cases by reviewing the current reports from the literature.</p><p><strong>Methods: </strong>A 24-year-old woman was admitted to the outpatient endocrinology unit with a diagnosis of primary amenorrhea, history of Growth Hormone deficiency and multiple congenital anomalies including rectal atresia. The subsequent hormonal investigation led to the diagnosis of hypogonadotropic hypogonadism and persistent GH deficiency. Abdominal and pelvic ultrasounds were normal whereas the brain MRI revealed a hypoplastic sella turcica with a hypoplastic anterior pituitary lobe, an ectopic posterior pituitary lobe and a thin pituitary stalk. The genetic analysis revealed a novel pathogenic missense heterozygous variant (c.1958G > A, p.Agr635Gln) in exon 15 of FGFR1 gene. PubMed, Scopus, and Web of Science were searched for the identification of studies reporting cases of CPHD with FGFR1 gene defects.</p><p><strong>Results: </strong>Of the 648 records retrieved, 10 were included in this review. A comprehensive overview of the cases was summarized, and their clinical and genetic characteristics were presented.</p><p><strong>Conclusion: </strong>Although FGFR1 variants have been associated with Kallmann syndrome and isolated hypogonadotropic hypogonadism and recently with CPHD, the patient's phenotype includes phenotypic alterations not previously described, to the best of our knowledge, within the spectrum of non-reproductive features of either of these entities. Isolated GH deficiency combined with other non-common abnormalities exerts a great possibility for subsequent CPHD manifestation.</p>","PeriodicalId":49211,"journal":{"name":"Endocrine","volume":" ","pages":"556-564"},"PeriodicalIF":2.9000,"publicationDate":"2025-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12289763/pdf/","citationCount":"0","resultStr":"{\"title\":\"A rare combination of hypogonadotropic hypogonadism, GH deficiency and rectal atresia in a female with an FGFR1 variant: a case report and systematic review of the literature.\",\"authors\":\"Krystallenia I Alexandraki, Odysseas Violetis, Eleni Memi, Helen Fryssira, Vasileios Papanikolaou, Maria Papagianni, George Mastorakos\",\"doi\":\"10.1007/s12020-025-04261-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Purpose: </strong>To report a case with combined pituitary hormone deficiency (CPHD) and Fibroblast growth factor receptor 1 (FGFR1) gene defect, and summarize the clinical characteristics of similar cases by reviewing the current reports from the literature.</p><p><strong>Methods: </strong>A 24-year-old woman was admitted to the outpatient endocrinology unit with a diagnosis of primary amenorrhea, history of Growth Hormone deficiency and multiple congenital anomalies including rectal atresia. The subsequent hormonal investigation led to the diagnosis of hypogonadotropic hypogonadism and persistent GH deficiency. Abdominal and pelvic ultrasounds were normal whereas the brain MRI revealed a hypoplastic sella turcica with a hypoplastic anterior pituitary lobe, an ectopic posterior pituitary lobe and a thin pituitary stalk. The genetic analysis revealed a novel pathogenic missense heterozygous variant (c.1958G > A, p.Agr635Gln) in exon 15 of FGFR1 gene. PubMed, Scopus, and Web of Science were searched for the identification of studies reporting cases of CPHD with FGFR1 gene defects.</p><p><strong>Results: </strong>Of the 648 records retrieved, 10 were included in this review. A comprehensive overview of the cases was summarized, and their clinical and genetic characteristics were presented.</p><p><strong>Conclusion: </strong>Although FGFR1 variants have been associated with Kallmann syndrome and isolated hypogonadotropic hypogonadism and recently with CPHD, the patient's phenotype includes phenotypic alterations not previously described, to the best of our knowledge, within the spectrum of non-reproductive features of either of these entities. Isolated GH deficiency combined with other non-common abnormalities exerts a great possibility for subsequent CPHD manifestation.</p>\",\"PeriodicalId\":49211,\"journal\":{\"name\":\"Endocrine\",\"volume\":\" \",\"pages\":\"556-564\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-08-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12289763/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Endocrine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s12020-025-04261-4\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/5/28 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Endocrine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s12020-025-04261-4","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/5/28 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
摘要
目的:报告1例垂体激素缺乏症(CPHD)合并成纤维细胞生长因子受体1 (FGFR1)基因缺陷,并通过回顾目前文献报道,总结类似病例的临床特点。方法:一名24岁的女性以原发性闭经、生长激素缺乏史和直肠闭锁等多种先天性异常就诊于门诊内分泌科。随后的激素调查导致诊断为促性腺功能低下和持续GH缺乏。腹部和盆腔超声检查正常,而脑部MRI显示蝶鞍发育不全,垂体前叶发育不全,垂体后叶异位,垂体柄薄。遗传分析显示,在FGFR1基因的第15外显子中存在一种新的致病性错义杂合变异(c.1958G > a, p.Agr635Gln)。检索PubMed、Scopus和Web of Science以确定报告FGFR1基因缺陷的CPHD病例的研究。结果:在检索到的648条记录中,有10条被纳入本综述。本文对病例进行了综述,并对其临床和遗传学特征进行了介绍。结论:尽管FGFR1变异与Kallmann综合征和孤立性促性腺功能减退症以及最近与CPHD有关,但据我们所知,患者的表型包括以前未描述的表型改变,在这两种实体的非生殖特征范围内。孤立的生长激素缺乏合并其他不常见的异常,为随后的CPHD表现提供了很大的可能性。
A rare combination of hypogonadotropic hypogonadism, GH deficiency and rectal atresia in a female with an FGFR1 variant: a case report and systematic review of the literature.
Purpose: To report a case with combined pituitary hormone deficiency (CPHD) and Fibroblast growth factor receptor 1 (FGFR1) gene defect, and summarize the clinical characteristics of similar cases by reviewing the current reports from the literature.
Methods: A 24-year-old woman was admitted to the outpatient endocrinology unit with a diagnosis of primary amenorrhea, history of Growth Hormone deficiency and multiple congenital anomalies including rectal atresia. The subsequent hormonal investigation led to the diagnosis of hypogonadotropic hypogonadism and persistent GH deficiency. Abdominal and pelvic ultrasounds were normal whereas the brain MRI revealed a hypoplastic sella turcica with a hypoplastic anterior pituitary lobe, an ectopic posterior pituitary lobe and a thin pituitary stalk. The genetic analysis revealed a novel pathogenic missense heterozygous variant (c.1958G > A, p.Agr635Gln) in exon 15 of FGFR1 gene. PubMed, Scopus, and Web of Science were searched for the identification of studies reporting cases of CPHD with FGFR1 gene defects.
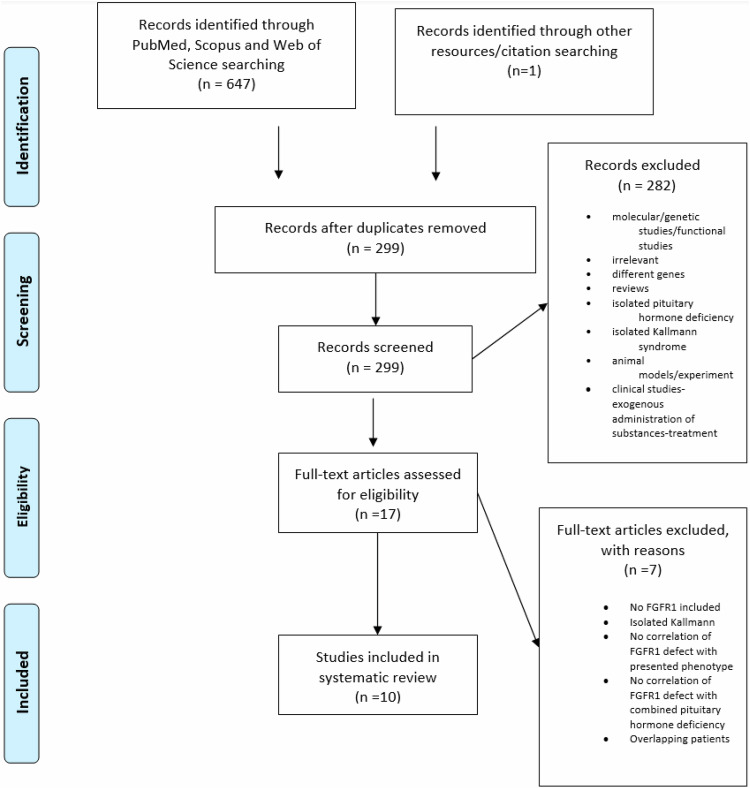
Results: Of the 648 records retrieved, 10 were included in this review. A comprehensive overview of the cases was summarized, and their clinical and genetic characteristics were presented.
Conclusion: Although FGFR1 variants have been associated with Kallmann syndrome and isolated hypogonadotropic hypogonadism and recently with CPHD, the patient's phenotype includes phenotypic alterations not previously described, to the best of our knowledge, within the spectrum of non-reproductive features of either of these entities. Isolated GH deficiency combined with other non-common abnormalities exerts a great possibility for subsequent CPHD manifestation.
期刊介绍:
Well-established as a major journal in today’s rapidly advancing experimental and clinical research areas, Endocrine publishes original articles devoted to basic (including molecular, cellular and physiological studies), translational and clinical research in all the different fields of endocrinology and metabolism. Articles will be accepted based on peer-reviews, priority, and editorial decision. Invited reviews, mini-reviews and viewpoints on relevant pathophysiological and clinical topics, as well as Editorials on articles appearing in the Journal, are published. Unsolicited Editorials will be evaluated by the editorial team. Outcomes of scientific meetings, as well as guidelines and position statements, may be submitted. The Journal also considers special feature articles in the field of endocrine genetics and epigenetics, as well as articles devoted to novel methods and techniques in endocrinology.
Endocrine covers controversial, clinical endocrine issues. Meta-analyses on endocrine and metabolic topics are also accepted. Descriptions of single clinical cases and/or small patients studies are not published unless of exceptional interest. However, reports of novel imaging studies and endocrine side effects in single patients may be considered. Research letters and letters to the editor related or unrelated to recently published articles can be submitted.
Endocrine covers leading topics in endocrinology such as neuroendocrinology, pituitary and hypothalamic peptides, thyroid physiological and clinical aspects, bone and mineral metabolism and osteoporosis, obesity, lipid and energy metabolism and food intake control, insulin, Type 1 and Type 2 diabetes, hormones of male and female reproduction, adrenal diseases pediatric and geriatric endocrinology, endocrine hypertension and endocrine oncology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


